Quinolinic acid
 | |
| Names | |
|---|---|
| IUPAC name
Pyridine-2,3-dicarboxylic acid | |
| Identifiers | |
| 89-00-9 | |
| 3D model (Jmol) | Interactive image |
| ChEBI | CHEBI:16675 |
| ChEMBL | ChEMBL286204 |
| ChemSpider | 1037 |
| ECHA InfoCard | 100.001.704 |
| EC Number | 201-874-8 |
| KEGG | C03722 |
| MeSH | D017378 |
| PubChem | 1066 |
| |
| |
| Properties | |
| C7H5NO4 | |
| Molar mass | 167.12 g/mol |
| Melting point | 185 to 190 °C (365 to 374 °F; 458 to 463 K) (decomposes) |
| Hazards | |
| Safety data sheet | External MSDS |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| | |
| Infobox references | |
Quinolinic acid (abbreviated QUIN or QA), also known as pyridine-2,3-dicarboxylic acid, is a dicarboxylic acid with a pyridine backbone. It is a colorless solid. It is the biosynthetic precursor to nicotine.[1]
Quinolinic acid is a downstream product of the kynurenine pathway, which metabolizes the amino acid tryptophan. It acts as an NMDA receptor agonist.[2]
Quinolinic acid has a potent neurotoxic effect. Studies have demonstrated that quinolinic acid may be involved in many psychiatric disorders, neurodegenerative processes in the brain, as well as other disorders. Within the brain, quinolinic acid is only produced by activated microglia and macrophages.[3]
History
In 1949 L. Henderson was one of the earliest to describe quinolinic acid. Lapin followed up this research by demonstrating that quinolinic acid could induce convulsions when injected into mice brain ventricles. However, it was not until 1981 that Stone and Perkins showed that quinolinic acid activates the N-methyl-d-aspartate receptor (NMDAR). After this, Schwarcz demonstrated that elevated quinolinic acid levels could lead to axonal neurodegeneration.[4]
Synthesis
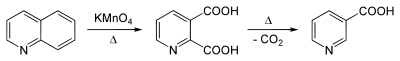
One of the earliest reported syntheses of this quinolinic acid was by Zdenko Hans Skraup, who found that methyl-substituted quinolines could be oxidized to quinolinic acid by potassium permanganate.[5]
This compound is commercially available. It is generally obtained by the oxidation of quinoline. Oxidants such as ozone,[6] hydrogen peroxide,[7] and potassium permanganate have been used. Electrolysis is able to perform the transformation as well.[8][9]
Quinolinic acid may undergo further decarboxylation to nicotinic acid (niacin):
Biosynthesis
From aspartate
Oxidation of aspartate by the enzyme aspartate oxidase gives iminosuccinate, containing the two carboxylic acid groups that are found in quinolinic acid. Condensation of iminosuccinate with glyceraldehyde-3-phosphate, mediated by quinolinate synthase, affords quinolinic acid.[1]
Catabolism of tryptophan
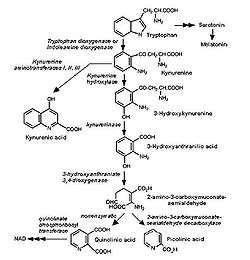
Quinolinic acid is a byproduct of the kynurenine pathway, which is responsible for catabolism of tryptophan in mammals. This pathway is important for its production of the coenzyme nicotinamide adenine dinucleotide (NAD+) and produces several neuroactive intermediates including quinolinic acid, kynurenine (KYN), kynurenic acid (KYNA), 3-hydroxykynurenine (3-HK), and 3-hydroxyanthranilic acid (3-HANA).[10][11] Quinolinic acid's neuroactive and excitatory properties are a result of NMDA receptor agonism in the brain.[11] It also acts as a neurotoxin, gliotoxin, proinflammatory mediator, and pro-oxidant molecule.[10]
Quinolinic acid is unable to pass through the blood-brain barrier (BBB) and must be produced within the brain microglial cells or macrophages that have passed the BBB.[10] While quinolinic acid cannot pass the BBB, kynurenic acid, tryptophan and 3-hydroxykynurenine do and subsequently act as precursors to the production of quinolinic acid in the brain. The quinolinic acid produced in microglia is then released and stimulates NMDA receptors resulting in excitatory neurotoxiticity.[11] While astrocytes are not able to produce quinolinic acid directly, they are capable of producing KYNA, which when released from the astrocytes can be taken in by migroglia that can in turn increase quinolinic acid production.[10][11]
Microglia and macrophages produce the vast majority of quinolinic acid present in the body. This production is increased during an immune response. It is suspected that this is a result of activation of indoleamine dioxygenases (to be specific, IDO-1 and IDO-2) as well as tryptophan 2,3-dioxygenase (TDO) stimulation by inflammatory cytokines (mainly IFN-gamma, but also IFN-beta and IFN-alpha).[10]
IDO-1, IDO-2 and TDO are present in microglia and macrophages. Under inflammatory conditions and conditions of T cell activation, leukocytes are retained in the brain by cytokine and chemokine production, which can lead to the breakdown of the BBB, thus increasing the quinolinic acid that enters the brain. Furthermore, quinolinic acid has been shown to play a role in destabilization of the cytoskeleton within astrocytes and brain endothelial cells, contributing to the degradation of the BBB, which results in higher concentrations of quinolinic acid in the brain.[12]
Toxicity
Quinolinic acid is an excitotoxin in the CNS. It reaches pathological levels in response to inflammation in the brain, which activates resident microglia and macrophages. High levels of quinolinic acid can lead to hindered neuronal function or even apoptotic death.[10] Quinolinic acid produces its toxic effect through several mechanisms, primarily as its function as an NMDA receptor agonist, which triggers a chain of deleterious effects, but also through lipid peroxidation, and cytoskeletal destabilization.[10] The gliotoxic effects of quinolinic acid further amplify the inflammatory response. Quinolinic acid affects neurons located mainly in the hippocampus, striatum, and neocortex, due to the selectivity toward quinolinic acid by the specific NMDA receptors residing in those regions.[10]
When inflammation occurs, quinolinic acid is produced in excessive levels through the kynurenine pathway. This leads to over excitation of the NMDA receptor, which results in an influx of Ca2+ into the neuron. High levels of Ca2+ in the neuron trigger an activation of destructive enzymatic pathways including protein kinases, phospholipases, NO synthase, and proteases.[13] These enzymes will degenerate crucial proteins in the cell and increase NO levels, leading to an apoptotic response by the cell, which results in cell death.
In normal cell conditions, astrocytes in the neuron will provide a glutamate-glutamine cycle, which results in reuptake of glutamate from the synapse into the pre-synaptic cell to be recycled, keeping glutamate from accumulating to lethal levels inside the synapse. At high concentrations, quinolinic acid inhibits glutamine synthetase, a critical enzyme in the glutamate-glutamine cycle. In addition, It can also promote glutamate release and block its reuptake by astrocytes. All three of these actions result in increased levels of glutamate activity that could be neurotoxic.[10]
This results in a loss of function of the cycle, and results in an accumulation of glutamate. This glutamate further stimulates the NMDA receptors, thus acting synergistically with quinolinic acid to increase its neurotoxic effect by increasing the levels of glutamate, as well as inhibiting its uptake. In this way, quinolinic acid self-potentiates its own toxicity.[10] Furthermore, quinolinic acid results in changes of the biochemistry and structure of the astrocytes themselves, resulting in an apoptotic response. A loss of astrocytes results in a pro-inflammatory effect, further increasing the initial inflammatory response which initiates quinolinic acid production.[10]
Quinolinic acid can also exert neurotoxicity through lipid peroxidation, as a result of its pro-oxidant properties. Quinolinic acid can interact with Fe(II) to form a complex that induces a reactive oxygen and nitrogen species (ROS/RNS), notably the hydroxyl radical •OH. This free radical causes oxidative stress by further increasing glutamate release and inhibiting its reuptake, and results in the breakdown of DNA in addition to lipid peroxidation.[13] Quinolinic acid has also been noted to increase phosphorylation of proteins involved in cell structure, leading to destabilization of the cytoskeleton.[10]
Clinical implications
Psychiatric disorders
Mood disorders
The prefrontal cortices in the post-mortem brains of patients with major depression and bipolar depression contain increased levels quinolinic acid immunoreactivity compared to the brains of patients never having suffered from depression.[14] The fact that NMDA receptor antagonists possess antidepressant properties suggests that increased levels of quinolinic acid in patients with depression may overactivate NMDA receptors.[11] By inducing increased levels of quinolinic acid in the cerebral spinal fluid with interferon α, researchers have demonstrated that increased quinolinic acid levels correlate with increased depressive symptoms.[15]
Increased levels of quinolinic acid might contribute to the apoptosis of astrocytes and certain neurons, resulting in decreased synthesis of neurotrophic factors. With less neurotrophic factors, the astrocyte-microglia-neuronal network is weaker and thus is more likely to be affected by environmental factors such as stress. In addition, increased levels of quinolinic acid could play a role in impairment of the glial-neuronal network, which could be associated with the recurrent and chronic nature of depression.[14]
Furthermore, studies have shown that unpredictable chronic mild stress (UCMS) can lead to the metabolism of quinolinic acid in the amygdala and striatum and a reduction in quinolinic acid pathway in the cingulate cortex. Experiments with mice demonstrate how quinolinic acid can affect behavior and act as endogenous anxiogens. For instance, when quinolinic acid levels are increased, mice socialize and groom for shorter periods of time.[15] There is also evidence that increased concentrations of quinolinic acid can play a role in adolescent depression.[14]
Schizophrenia
Quinolinic acid may be involved in schizophrenia; however, there has been no research done to examine the specific effects of quinolinic acid in schizophrenia. There are many studies that show that kynurenic acid (KYNA) plays a role in the positive symptoms of schizophrenia, and there has been some research to suggest that 3-hydroxykynurenine (OHK) plays a role in the disease as well. Because quinolinic acid is strongly associated with KYNA and OHK, it may too play a role in schizophrenia.[11][14]
Conditions related to neuronal death
The cytotoxic effects of quinolinic acid elaborated upon in the toxicity section amplifiy cell death in neurodegenerative conditions.
Amyotrophic lateral sclerosis (ALS)
Quinolinic acid may contribute to the causes of amyotrophic lateral sclerosis (ALS). Researchers have found elevated levels of quinolinic acid in the cerebral spinal fluid (CSF), motor cortex, and spinal cord in ALS patients. These increased concentrations of quinolinic acid could lead to neurotoxicity. In addition, quinolinic acid is associated with overstimulating NMDA receptors on motor neurons. Studies have demonstrated that quinolinic acid leads to depolarization of spinal motor neurons by interacting with the NMDA receptors on those cells in rats. Also, quinolinic acid plays a role in mitochondrial dysfunction in neurons. All of these effects could contribute to ALS symptoms.[16]
Alzheimer's disease
Researchers have found a correlation between quinolinic acid and Alzheimer's disease. For example, studies have found in the post-mortem brains of Alzheimer’s disease patients higher neuronal quinolinic acid levels and that quinolinic acid can associate with tau protein.[11][17] Furthermore, researchers have demonstrated that quinolinic acid increases tau phosphorylation in vitro in human fetal neurons [11][17] and induces ten neuronal genes including some known to correlate with Alzheimer’s disease.[17] In immunoreactivity studies, researchers have found that quinolinic acid immunoreactivity is strongest in glial cells that are located close to amyloid plaques and that there is immunoreactivity with neurofibrillary tangles.[11]
Brain ischemia
Brain ischemia is characterized by insufficient blood flow to the brain. Studies with ischaemic gerbils indicate that, after a delay, levels of quinolinic acid significantly increase, which correlates with increased neuronal damage.[14][18] In addition, researchers have found that, after transient global ischaemia, there are microglia containing quinolinic acid within the brain. Following cerebral ischaemia, delayed neuronal death may occur in part because of central microglia and macrophages, which possess and secrete quinolinic acid. This delayed neurodegeneration could be associated with chronic brain damage that follows a stroke.[18]
Human immunodeficiency virus (HIV) and Acquired immunodeficiency syndrome (AIDS)
Studies have found that there is a correlation between levels of quinolinic acid in cerebral spinal fluid (CSF) and HIV-associated neurocognitive disorder (HAND) severity. About 20% of HIV patients suffer from this disorder. Concentrations of quinolinic acid in the CSF are associated to different stages of HAND. For example, raised levels of quinolinic acid after infection are correlated to perceptual-motor slowing in patients. Then, in later stages of HIV, increased concentrations of quinolinic acid in the CSF of HAND patients correlates with HIV encephalitis and cerebral atrophy.[19]
Quinolinic acid has also been found in HAND patients’ brains. In fact, the amount of quinolinic acid found in the brain of HAND patients can be up to 300 times greater than that found in the CSF.[20] Neurons exposed to quinolinic acid for long periods of time can develop cytoskeletal abnormalities, vacuolization, and cell death. HAND patients’ brains contain many of these defects. Furthermore, studies in rats have demonstrated that quinolinic acid can lead to neuronal death in brains structures that are affected by HAND, including the striatum, hippocampus, the substantia nigra, and non-limbic cortex.[19]
Levels of quinolinic acid in the CSF of AIDS patients suffering from AIDS- dementia can be up to twenty times higher than normal. Similar to HIV patients, this increased quinolinic acid concentration correlates with cognitive and motor dysfunction. When patients were treated with zidovudine to decrease quinolinic acid levels, the amount of neurological improvement was related to the amount of quinolinic acid decreased.[20]
Huntington's disease
In the initial stages of Huntington's disease, patients have substantially increased quinolinic acid levels, in particular in the neostriatum and cortex. These areas of the brain that suffer the most damage at these stages.[16][18] The increase in quinolinic acid correlates with the early activation of microglia and increased cerebral 3-hydroxykynurenine (3-HK) levels. Furthermore, these increased levels of quinolinic acid are great enough to produce excitotoxic neuronal damage.[11] Studies have demonstrated that activation of NMDA receptors by quinolinic acid leads to neuronal dysfunction and death of striatal GABAergic medium spiny neurons (MSN).[16]
Researchers utilize quinolinic acid in order to study Huntington’s disease in many model organisms. Because injection of quinolinic acid into the striatum of rodents induces electrophysiological, neuropathological, and behavioral changes similar to those found in Huntington’s disease, this is the most common method researchers use to produce a Huntington’s disease phenotype.[14][18] Neurological changes produced by quinolinic acid injections include altered levels of glutamate, GABA, and other amino acids. Lesions in the pallidum can suppress effects of quinolinic acid in monkeys injected with quinolinic acid into their striatum. In humans, such lesions can also diminish some of the effects of Huntington’s disease and Parkinson's disease.[20]
Parkinson's disease
Quinolinic acid neurotoxicity is thought to play a role in Parkinson's disease.[16][21] Studies show that quinolinic acid is involved in the degeneration of the dopaminergic neurons in the substantia nigra (SN) of Parkinson's disease patients. SN degeneration is one of the key characteristics of Parkinson's disease. Microglia associated with dopaminergic cells in the SN produce quinolinic acid at this location when scientists induce Parkinson's disease symptoms in macaques. Quinolinic acid levels are too high at these sites to be controlled by KYNA, causing neurotoxicity to occur.[16]
Other
Quinolinic acid levels are increased in the brains of children infected with a range of bacterial infections of the central nervous system (CNS),[18][20] of poliovirus patients,[20] and of Lyme disease with CNS involvement patients.[14][20] In addition, raised quinolinic acid levels have been found in traumatic CNS injury patients, patients suffering from cognitive decline with ageing, hyperammonaemia patients, hypoglycaemia patients, and systemic lupus erythematosus patients. Also, it has been found that people suffering from malaria and patients with olivopontocerebellar atrophy have raised quinolinic acid metabolism.[20]
Treatment focus
Reduction of the excitotoxic effects of quinolinic acid is the subject of on-going research. NMDAr antagonists have been shown to provide protection to motor neurons from excitotoxicity resulting from quinolinic acid production.[10] Kyurenic acid, another product of the kyurenine pathway acts as an NMDA receptor antagonist.[22]
Kyurenic acid thus acts as a neuroprotectant, by reducing the dangerous over-activation of the NMDA receptors. Manipulation of the kyurenine pathway away from quinolinic acid and toward kyurenic acid is therefore a major therapeutic focus. Nicotinylalanine has been shown to be an inhibitor of kynurenine hydroxylase, which results in a decreased production of quinolinic acid, thus favoring kyurenic acid production.[22] This change in balance has the potential to reduce hyperexcitability, and thus excitotoxic damage produced from elevated levels of quinolinic acid.[22] Therapeutic efforts are also focusing on antioxidants, which have been shown to provide protection against the pro-oxidant properties of quinolinic acid.[10]
Norharmane suppresses the production of quinolinic acid, 3-hydroxykynurenine and nitric oxide synthase, thereby acting as a neuroprotectant.[23] Natural phenols such as catechin hydrate, curcumin, and epigallocatechin gallate reduce the neurotoxicity of quinolinic acid, via anti-oxidant and possibly calcium influx mechanisms.[24] COX-2 inhibitors, such as licofelone have also demonstrated protective properties against the neurotoxic effects of quinolinic acid. COX-2 is upregulated in many neurotoxic disorders and is associated with increased ROS production. Inhibitors have demonstrated some evidence of efficacy in mental health disorders such as major depressive disorder, schizophrenia, and Huntington's disease.[22]
See also
References
- 1 2 Hiroshi Ashihara, Alan Crozier, Atsushi Komamine "Nicotine Biosynthesis" in Plant Metabolism and Biotechnology, Tsubasa Shoji, Takashi Hashimoto Eds. Wiley-VCH, Weinheim, 2011. {{DOI: 10.1002/9781119991311.ch7}}
- ↑ Misztal M, Frankiewicz T, Parsons CG, Danysz W (January 1996). "Learning deficits induced by chronic intraventricular infusion of quinolinic acid--protection by MK-801 and memantine". Eur. J. Pharmacol. 296 (1): 1–8. doi:10.1016/0014-2999(95)00682-6. PMID 8720470.
- ↑ Guillemin, G.; Smith, Danielle G.; Smythe, George A.; Armati, Patricia J.; Brew, George J. (2003). "Expression of the kynurenine pathway enzymes in human microglia and macrophages". Adv Exp Med Biol. Advances in Experimental Medicine and Biology. 527: 105–12. doi:10.1007/978-1-4615-0135-0_12. ISBN 978-0-306-47755-3. PMID 15206722.
- ↑ Guillemin, Gilles J (March 2012). "Quinolinic acid: neurotoxicity". FEBS Journal. 279 (8): 1355. doi:10.1111/j.1742-4658.2012.08493.x. PMID 22251552.
- ↑ Skraup, Zd. H. (1881). "Synthetische Versuche in der Chinolinreihe". Monatshefte fü Chemie. 2: 139. doi:10.1007/BF01516502.
- ↑ WO 2010011134, H. Bruno, "Ozonolysis of Aromatics and/or Olefins"
- ↑ US Patent 4420616, Ikegami, Seishi & Hatano, Yoshihiro, "Oxidative process for the preparation of copper quinolinate", assigned to Yamamoto Kagaku Gosei KK
- ↑ Marshall Kulka (1946). "Electrolytic Oxidation of Quinoline and 3-Picoline". J. Am. Chem. Soc. 68 (12): 2472. doi:10.1021/ja01216a008. PMID 20282382.
- ↑ EP 0159769, Toomey Jr., Joseph E., "Electrochemical oxidation of pyridine bases", assigned to Reilly Industries, Inc.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Guillemin, Giles (April 2012). "Quinolinic acid, the inescapable neurotoxin". FEBS Journal. 279 (8): 1356–1365. doi:10.1111/j.1742-4658.2012.08485.x. PMID 22248144.
- 1 2 3 4 5 6 7 8 9 10 Schwarcz, Robert; John P. Bruno; Paul J. Muchowski; Hui-Qiu Wu (July 2012). "Kynurenines in the mammalian brain: when physiology meets pathology". Nature Reviews Neuroscience. 13 (7): 465–477. doi:10.1038/nrn3257. PMID 22678511.
- ↑ Combes, Valery; Gilles J. Guillemin; Tailoi Chan-Ling; Nicholas H. Hunt; Georges E.R. Grau (August 2012). "The crossroads of neuroinflammation in infectious diseases: endothelial cells and astrocytes". Trends in Parasitology. 28 (8): 311–319. doi:10.1016/j.pt.2012.05.008. PMID 22727810.
- 1 2 Pérez-De La Cruz, V.; P.Carrillo-Mora, A. Santamaría (2012). "Quinolinic acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms". International Journal of Tryptophan Research (5): 1–8. doi:10.4137/IJTR.S8158.
- 1 2 3 4 5 6 7 Myint, Aye M. (April 2012). "Kynurenines: from the perspective of major psychiatric disorders.". FEBS Journal. 279 (8): 1375–1385. doi:10.1111/j.1742-4658.2012.08551.x. PMID 22404766.
- 1 2 Maes, M; Leonard, B.E.; Myint, A.M.; Kubera, M.; Verkerk, R. (29 April 2011). "The new '5-HT' hypothesis of depression: Cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression". Progress in Neuro-Psychopharmacology & Biological Psychiatry. 35 (3): 702–721. doi:10.1016/j.pnpbp.2010.12.017. PMID 21185346.
- 1 2 3 4 5 Tan, Lin; Yu, JT; Tan, L (15 December 2012). "The kynurenine pathway in neurodegenerative diseases: Mechanistic and therapeutic considerations". Journal of the Neurological Sciences. 323 (1–2): 1–8. doi:10.1016/j.jns.2012.08.005. PMID 22939820.
- 1 2 3 Severino, Patricia Cardoso; Muller, GDS; Vandresen-Filho, S; Tasca, CI (10 October 2011). "Cell signaling in NMDA preconditioning and neuroprotection in convulsions induced by quinolinic acid". Life Sciences. 89 (15–16): 570–576. doi:10.1016/j.lfs.2011.05.014. PMID 21683718.
- 1 2 3 4 5 Stone, Trevor W.; Forrest, C.M.; Darlington, L.G. (April 2012). "Kynurenine pathway inhibition as a therapeutic strategy for neuroprotection". FEBS Journal. 279 (8): 1386–1397. doi:10.1111/j.1742-4658.2012.08487.x. PMID 22248239.
- 1 2 Kandanearatchi, Apsara; Brew, Bruce J. (April 2012). "The kynurenine pathway and quinolinic acid: pivotal roles in HIV associated neurocognitive disorders". FEBS Journal. 279 (8): 1366–1374. doi:10.1111/j.1742-4658.2012.08500.x. PMID 22260426.
- 1 2 3 4 5 6 7 Stone, Trevor W. (January 2001). "Endogenous neurotoxins from tryptophan". Toxicon. 39 (1): 61–73. doi:10.1016/S0041-0101(00)00156-2. PMID 10936623.
- ↑ Szabó, Nikoletta; Kincses, Z.T.; Toldi, J; Vécsei, L (15 November 2011). "Altered tryptophan metabolism in Parkinson's disease: A possible novel therapeutic approach". Journal of the Neurological Sciences. 310 (1-2): 256–260. doi:10.1016/j.jns.2011.07.021. PMID 21824629.
- 1 2 3 4 Kalonia, H.; P. Kumar; A. Kumar (2011). "Licofelone attenuates quinolinic acid induced huntington like symptoms: Possible behavioral, biochemical and cellular alterations". Progress in Neuro-Psychopharmacology & Biological Psychiatry. 35 (35(2)): 607–615. doi:10.1016/j.pnpbp.2011.01.003.
- ↑ Oxenkrug, Gregory F. (January 2011). "Interferon-gamma-inducible kynurenines/pteridines inflammation cascade: implications for aging and aging-associated psychiatric and medical disorders". Journal of Neural Transmission. 118 (1): 75–85. doi:10.1007/s00702-010-0475-7.
- ↑ Braidy N, Grant R, Adams S, Guillemin GJ (January 2010). "Neuroprotective effects of naturally occurring polyphenols on quinolinic acid-induced excitotoxicity in human neurons". FEBS J. 277 (2): 368–82. doi:10.1111/j.1742-4658.2009.07487.x. PMID 20015232.