Point mutation

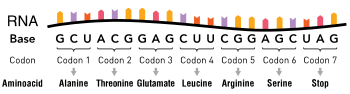
A point mutation, or single base modification, is a type of mutation that causes a single nucleotide base substitution, insertion, or deletion of the genetic material, DNA or RNA. The term frameshift mutation indicates the addition or deletion of a base pair.
A point mutant is an individual that is affected by a point mutation.
Repeat induced point mutations are recurring point mutations, discussed below.
Causes of point mutations
Point mutation is a random SNP (single-nucleotide polymorphism) mutation in the deoxyribonucleic acid (DNA) that occurs at one point. Point mutations usually take place during DNA replication. DNA replication occurs when one double-stranded DNA molecule creates two single strands of DNA, each of which is a template for the creation of the complementary strand. A single point mutation can change the whole DNA sequence. Changing one purine or pyrimidine may change the amino acid that the nucleotides code for.
Point mutations may arise from spontaneous mutations that occur during DNA replication. The rate of mutation may be increased by mutagens. Mutagens can be physical, such as radiation from UV rays, X-rays or extreme heat, or chemical (molecules that misplace base pairs or disrupt the helical shape of DNA). Mutagens associated with cancers are often studied to learn about cancer and its prevention.
There are multiple ways for point mutations to occur. First, ultraviolet (UV) light and higher-frequency light are capable of ionizing electrons, which in turn can affect DNA. Reactive oxygen molecules with free radicals, which are a byproduct of cellular metabolism, can also be very harmful to DNA. These reactants can lead to both single-stranded DNA breaks and double-stranded DNA breaks. Third, bonds in DNA eventually degrade, which creates another problem to keep the integrity of DNA to a high standard. There can also be replication errors that lead to substitution, insertion, or deletion mutations.
It was previously believed that these mutations happened completely by chance, with no regard for their effects on the organisms. Recently, there have been studies suggesting that these mutations occur in response to environmental challenges. That is to say, they are more likely to occur when they are advantageous to the organism, rather than when they are neutral or disadvantageous. When cells were deprived of a certain amino acid, tryptophan, for prolonged periods of time, point mutations in trp operon reverted to tryptophan, leading to an advantageous result, more frequently than under normal conditions when the mutations were neutral. In addition, the tryptophan mutation rate was unaffected when the cells were deprived of another amino acid, cysteine, further suggesting that the mutation rate was specific to situations in which the mutation was advantageous.[1]
Categorizing point mutations
Transition/Transversion categorization
In 1959 Ernst Freese coined the terms "transitions" or "transversions" to categorize different types of point mutations.[2][3]
- Transitions: replacement of a purine base with another purine or replacement of a pyrimidine with another pyrimidine
- Transversions: replacement of a purine with a pyrimidine or vice versa.

There is a systematic difference in mutation rates for transitions (Alpha) and transversions (Beta). Transition mutations are about ten times more common than transversions.
Functional categorization
- Nonsense mutations: Code for a stop, which can truncate the protein. A nonsense mutation converts an amino acid codon into a termination codon. This causes the protein to be shortened because of the stop codon interrupting its normal code. How much of the protein is lost determines whether or not the protein is still functional.[4]
- Missense mutations: Code for a different amino acid. A missense mutation changes a codon so that a different protein is created, a non-synonymous change.[4]
- Conservative mutations: Result in an amino acid change. However, the properties of the amino acid remain the same (e.g., hydrophobic, hydrophilic, etc.). At times, a change to one amino acid in the protein is not detrimental to the organism as a whole. Most proteins can withstand one or two point mutations before their function changes.
- Non-conservative mutations: Result in an amino acid change that has different properties than the wild type. The protein may lose its function, which can result in a disease in the organism. For example, sickle-cell disease is caused by a single point mutation (a missense mutation) in the beta-hemoglobin gene that converts a GAG codon into GUG, which encodes the amino acid valine rather than glutamic acid. The protein may also exhibit a "gain of function" or become activated, such is the case with the mutation changing a valine to glutamic acid in the braf gene; this leads to an activation of the RAF protein which causes unlimited proliferative signalling in cancer cells.[5] These are both examples of a non-conservative (missense) mutation.
- Silent mutations: Code for the same amino acid. A silent mutation has no effect on the functioning of the protein. A single nucleotide can change, but the new codon specifies the same amino acid, resulting in an unmutated protein. This type of change is called synonymous change, since the old and new codon code for the same amino acid. This is possible because 64 codons specify only 20 amino acids. Different codons can lead to differential protein expression levels, however.[4]
Use of term to describe single base pair insertions and deletions
- Sometimes the term point mutation is used to describe insertions or deletions of a single base pair (which has more of an adverse effect on the synthesized protein due to the nucleotides' still being read in triplets, but in different frames: a mutation called a frameshift mutation).[4]
General consequences of point mutations
Point mutations that occur in non-coding sequences are most often without consequences, although there are exceptions. If the mutated base pair is in the promoter sequence of a gene, then the expression of the gene may change. Also, if the mutation occurs in the splicing site of an intron, then this may interfere with correct splicing of the transcribed pre-mRNA.
By altering just one amino acid, the entire peptide may change, thereby changing the entire protein. If the protein functions in cellular reproduction then this single point mutation can change the entire process of cellular reproduction for this organism.
Point germline mutations can lead to beneficial as well as harmful traits or diseases. This leads to adaptations based on the environment where the organism lives. An advantageous mutation can create an advantage for that organism and lead to the trait's being passed down from generation to generation, improving and benefiting the entire population. The scientific theory of evolution is greatly dependent on point mutations in cells. The theory explains the diversity and history of living organisms on Earth. In relation to point mutations, it states that beneficial mutations allow the organism to thrive and reproduce, thereby passing its positively affected mutated genes on to the next generation. On the other hand, harmful mutations cause the organism to die or be less likely to reproduce in a phenomenon known as natural selection.
There are different short-term and long-term effects that can arise from mutations. Smaller ones would be a halting of the cell cycle at numerous points. This means that a codon coding for the amino acid glycine may be changed to a stop codon, causing the proteins that should have been produced to be deformed and unable to complete their intended tasks. Because the mutations can affect the DNA and thus the chromatin, it can prohibit mitosis from occurring due to the lack of a complete chromosome. Problems can also arise during the processes of transcription and replication of DNA. These all prohibit the cell from reproduction and thus lead to the death of the cell. Long-term effects can be a permanent changing of a chromosome, which can lead to a mutation. These mutations can be either beneficial or detrimental. Cancer is an example of how they can be detrimental.[6]
Other effects of point mutations, or single nucleotide polymorphisms in DNA, depend on the location of the mutation within the gene. For example, if the mutation occurs in the region of the gene responsible for coding, the amino acid sequence of the encoded protein may be altered, causing a change in the function, activation, localization, or stability of the protein. Moreover, if the mutation occurs in the region of the gene where transcriptional machinery binds to the protein, the mutation can affect the binding of the transcription factors because the short nucleotide sequences recognized by the transcription factors will be altered. Mutations in this region can affect rate of efficiency of gene transcription, which in turn can alter levels of mRNA and, thus, protein levels in general.
Point mutations can have several effects on the behavior and reproduction of a protein depending on where the mutation occurs in the amino acid sequence of the protein. If the mutation occurs in the region of the gene that is responsible for coding for the protein, the amino acid may be altered. This slight change in the sequence of amino acids can cause a change in the function, activation of the protein meaning how it binds with a given enzyme, where the protein will be located within the cell, or the amount of free energy stored within the protein.
If the mutation occurs in the region of the gene where transcriptional machinery binds to the protein, the mutation can affect the way in which transcription factors bind to the protein. The mechanisms of transcription bind to a protein through recognition of short nucleotide sequences. A mutation in this region may alter these sequences and, thus, change the way the transcription factors bind to the protein. Mutations in this region can affect the efficiency of gene transcription, which controls both the levels of mRNA and overall protein levels.[7]
Specific diseases caused by point mutations
Cystic fibrosis
A defect in the cystic fibrosis transmembrane conductance regulator (CFTR) gene causes cystic fibrosis (CF). A protein made by this gene controls the movement of the water and salt in and out of the body's cells. Genes in people with CF incorrectly code proteins. This causes thick, sticky mucus and very salty sweat.[8][9]
Cancer
Point mutations in multiple tumor suppressor proteins cause cancer. For instance, point mutations in Adenomatous Polyposis Coli promote tumorigenesis.[10] A novel assay, Fast parallel proteolysis (FASTpp), might help swift screening of specific stability defects in individual cancer patients.[11]
Neurofibromatosis
Neurofibromatosis is caused by point mutations in the Neurofibromin 1[12][13] or Neurofibromin 2 gene.[14]
Sickle-cell anemia
Sickle-cell anemia is caused by a point mutation in the β-globin chain of hemoglobin, causing the hydrophilic amino acid glutamic acid to be replaced with the hydrophobic amino acid valine at the sixth position.
The β-globin gene is found on the short arm of chromosome 11. The association of two wild-type α-globin subunits with two mutant β-globin subunits forms hemoglobin S (HbS). Under low-oxygen conditions (being at high altitude, for example), the absence of a polar amino acid at position six of the β-globin chain promotes the non-covalent polymerisation (aggregation) of hemoglobin, which distorts red blood cells into a sickle shape and decreases their elasticity.[15]
Hemoglobin is a protein found in red blood cells, and is responsible for the transportation of oxygen through the body.[16] There are two subunits that make up the hemoglobin protein: beta-globins and alpha-globins.[17] Beta-hemoglobin is created from the genetic information on the HBB, or "hemoglobin, beta" gene found on chromosome 11p15.5.[18] A single point mutation in this polypeptide chain, which is 147 amino acids long, results in the disease known as Sickle Cell Anemia.[19] Sickle-Cell Anemia is an autosomal recessive disorder that affects 1 in 500 African Americans, and is one of the most common blood disorders in the United States.[18] The single replacement of the sixth amino acid in the beta-globin, glutamic acid, with valine results in deformed red blood cells. These sickle-shaped cells cannot carry nearly as much oxygen as normal red blood cells and they get caught more easily in the capillaries, cutting off blood supply to vital organs. The single nucleotide change in the beta-globin means that even the smallest of exertions on the part of the carrier results in severe pain and even heart attack. Below is a chart depicting the first thirteen amino acids in the normal and abnormal sickle cell polypeptide chain.[19]
Sequence for Normal Hemoglobin
| AUG | GUG | CAC | CUG | ACU | CCU | GAG | GAG | AAG | UCU | GCC | GUU | ACU |
| START | Val | His | Leu | Thr | Pro | Glu | Glu | Lys | Ser | Ala | Val | Thr |
Sequence for Sickle Cell Hemoglobin
| AUG | GUG | CAC | CUG | ACU | CCU | GUG | GAG | AAG | UCU | GCC | GUU | ACU |
| START | Val | His | Leu | Thr | Pro | Val | Glu | Lys | Ser | Ala | Val | Thr |
Tay–Sachs disease
The cause of Tay–Sachs disease is a genetic defect that is passed from parent to child. This genetic defect is located in the HEXA gene, which is found on chromosome 15.
The HEXA gene makes part of an enzyme called beta-hexosaminidase A, which plays a critical role in the nervous system. This enzyme helps break down a fatty substance called GM2 ganglioside in nerve cells. Mutations in the HEXA gene disrupt the activity of beta-hexosaminidase A, preventing the breakdown of the fatty substances. As a result, the fatty substances accumulate to deadly levels in the brain and spinal cord. The buildup of GM2 ganglioside causes progressive damage to the nerve cells. This is the cause of the signs and symptoms of Tay-Sachs disease.[20]
Color blindness
People who are colorblind have mutations in their genes that cause a loss of either red or green cones, and they therefore have a hard time distinguishing between colors. There are three kinds of cones in the human eye: red, green, and blue. Now researchers have discovered that some people with the gene mutation that causes colorblindness lose an entire set of "color" cones with no change to the clearness of their vision overall.[21]
Repeat induced point-mutation
In molecular biology, repeat induced point-mutation or RIP is a process by which DNA accumulates G:C to A:T transition mutations. Genomic evidence indicates that RIP occurs or has occurred in a variety of fungi[22] while experimental evidence indicates that RIP is active in Neurospora crassa,[23] Podospora anserina,[24] Magnaporthe grisea,[25] Leptosphaeria maculans,[26] Gibberella zeae[27] and Nectria haematococca.[28] In Neurospora crassa, sequences mutated by RIP are often methylated de novo.[23]
RIP occurs during the sexual stage in haploid nuclei after fertilization but prior to meiotic DNA replication.[23] In Neurospora crassa, repeat sequences of at least 400 base pairs in length are vulnerable to RIP. Repeats with as low as 80% nucleotide identity may also be subject to RIP. Though the exact mechanism of repeat recognition and mutagenesis are poorly understood, RIP results in repeated sequences undergoing multiple transition mutations.
The RIP mutations do not seem to be limited to repeated sequences. Indeed, for example, in the phytopathogenic fungus L. maculans, RIP mutations are found in single copy regions, adjacent to the repeated elements. These regions are either non-coding regions or genes encoding small secreted proteins including avirulence genes. The degree of RIP within these single copy regions was proportional to their proximity to repetitive elements.[29]
Rep and Kistler have speculated that the presence of highly repetitive regions containing transposons, may promote mutation of resident effector genes.[30] So the presence of effector genes within such regions is suggested to promote their adaptation and diversification when exposed to strong selection pressure.[31]
As RIP mutation is traditionally observed to be restricted to repetitive regions and not single copy regions, Fudal et al. [32] suggested that leakage of RIP mutation might occur within a relatively short distance of a RIP-affected repeat. Indeed, this has been reported in N. crassa whereby leakage of RIP was detected in single copy sequences at least 930 bp from the boundary of neighbouring duplicated sequences.[33] To elucidate the mechanism of detection of repeated sequences leading to RIP may allow to understand how the flanking sequences may also be affected.
Mechanism
RIP causes G:C to A:T transition mutations within repeats, however, the mechanism that detects the repeated sequences is unknown. RID is the only known protein essential for RIP. It is a DNA methyltransferease-like protein, that when mutated or knocked out results in loss of RIP.[34] Deletion of the rid homolog in Aspergillus nidulans, dmtA, results in loss of fertility[35] while deletion of the rid homolog in Ascobolus immersens, masc1, results in fertility defects and loss of methylation induced premeiotically (MIP).[36]
Consequences
RIP is believed to have evolved as a defense mechanism against Transposable Elements, which resemble parasites by invading and multiplying within the genome. RIP creates multiple missense and nonsense mutations in the coding sequence. This hypermutation of G-C to A-T in repetitive sequences eliminates functional gene products of the sequence (if there were any to begin with). In addition, many of the C-bearing nucleotides become methylated, thus decreasing transcription.
Use in molecular biology
Because RIP is so efficient at detecting and mutating repeats, fungal biologists often use it as a tool for mutagenesis. A second copy of a single-copy gene is first transformed into the genome. The fungus must then mate and go through its sexual cycle to activate the RIP machinery. Many different mutations within the duplicated gene are obtained from even a single fertilization event so that inactivated alleles, usually due to nonsense mutations, as well as alleles containing missense mutations can be obtained.[37]
History
The Cellular Reproduction process of Meiosis was discovered by Oscar Hertwig in 1876. Mitosis was discovered several years later in 1882 by Walther Flemming.
Hertwig studied sea urchins, and noticed that each egg contained one nucleus prior to fertilization and two nuclei after. This discovery proved that one spermatozoon could fertilize an egg, and therefore proved the process of meiosis. Hermann Fol continued Hertwig’s research by testing the effects of injecting several spermatozoa into an egg, and found that the process did not work with more than one spermatozoon.[38]
Flemming began his research of cell division starting in 1868. The study of cells was an increasingly popular topic in this time period. By 1873, Schneider had already begun to describe the steps of cell division. Flemming furthered this description in 1874 and 1875 as he explained the steps in more detail. He also argued with Schneider’s findings that the nucleus separated into rod-like structures by suggesting that the nucleus actually separated into threads that in turn separated. Flemming concluded that cells replicate through cell division, to be more specific mitosis.[39]
Matthew Meselson and Franklin Stahl are credited with the discovery of DNA replication. Watson and Crick acknowledged that the structure of DNA did indicate that there is some form of replicating process. However, there was not a lot of research done on this aspect of DNA until after Watson and Crick. People considered all possible methods of determining the replication process of DNA, but none were successful until Meselson and Stahl. Meselson and Stahl introduced a heavy isotope into some DNA and traced its distribution. Through this experiment, Meselson and Stahl were able to prove that DNA reproduces semi-conservatively.[40]
References
- ↑ Hall BG (September 1990). "Spontaneous point mutations that occur more often when advantageous than when neutral". Genetics. 126 (1): 5–16. PMC 1204135
 . PMID 2227388.
. PMID 2227388. - ↑ Freese, Ernst (April 1959). "The difference between spontaneous and base-analogue induced mutations of phage T4". Proc. Natl. Acad. Sci. U.S.A. 45 (4): 622–33. doi:10.1073/pnas.45.4.622. PMC 222607. PMID 16590424.
- ↑ Freese, Ernst (1959). "The Specific Mutagenic Effect of Base Analogues on Phage T4". J. Mol. Biol. 1 (2): 87–105. doi:10.1016/S0022-2836(59)80038-3.
- 1 2 3 4 http://members.cox.net/amgough/Fanconi-genetics-genetics-primer.htm
- ↑ Davies H, Bignell GR, Cox C, et al. (June 2002). "Mutations of the BRAF gene in human cancer". Nature. 417 (6892): 949–54. doi:10.1038/nature00766. PMID 12068308.
- ↑ Hoeijmakers JH (May 2001). "Genome maintenance mechanisms for preventing cancer". Nature. 411 (6835): 366–74. doi:10.1038/35077232. PMID 11357144.
- ↑ "A Shortcut to Personalized Medicine". Genetic Engineering & Biotechnology News. 18 June 2008.
- ↑ http://cystic-fibrosis.emedtv.com/cystic-fibrosis/causes-of-cystic-fibrosis.html
- ↑ http://www.nhlbi.nih.gov/health/dci/Diseases/cf/cf_causes.html
- ↑ Minde DP, Anvarian Z, Rüdiger SG, Maurice MM (2011). "Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer?". Mol. Cancer. 10: 101. doi:10.1186/1476-4598-10-101. PMC 3170638. PMID 21859464.
- ↑ Minde DP, Maurice MM, Rüdiger SG (2012). "Determining biophysical protein stability in lysates by a fast proteolysis assay, FASTpp". PLoS ONE. 7 (10): e46147. doi:10.1371/journal.pone.0046147. PMC 3463568. PMID 23056252.
- ↑ Serra, E; Ars, E; Ravella, A; Sánchez, A; Puig, S; Rosenbaum, T; Estivill, X; Lázaro, C (2001). "Somatic NF1 mutational spectrum in benign neurofibromas: MRNA splice defects are common among point mutations". Human Genetics. 108 (5): 416–29. PMID 11409870.
- ↑ Wiest, V; Eisenbarth, I; Schmegner, C; Krone, W; Assum, G (2003). "Somatic NF1 mutation spectra in a family with neurofibromatosis type 1: Toward a theory of genetic modifiers". Human Mutation. 22 (6): 423–7. doi:10.1002/humu.10272. PMID 14635100.
- ↑ Mohyuddin, A; Neary, W. J.; Wallace, A; Wu, C. L.; Purcell, S; Reid, H; Ramsden, R. T.; Read, A; Black, G; Evans, D. G. (2002). "Molecular genetic analysis of the NF2 gene in young patients with unilateral vestibular schwannomas". Journal of Medical Genetics. 39 (5): 315–22. doi:10.1136/jmg.39.5.315. PMC 1735110. PMID 12011146.
- ↑ http://www.ncbi.nlm.nih.gov/disease/sickle.html
- ↑ Hsia CC (January 1998). "Respiratory function of hemoglobin". N. Engl. J. Med. 338 (4): 239–47. doi:10.1056/NEJM199801223380407. PMID 9435331.
- ↑ "HBB — Hemoglobin, Beta". Genetics Home Reference. National Library of Medicine.
- 1 2 "Anemia, Sickle Cell". Genes and Disease. Bethesda MD: National Center for Biotechnology Information. 1998. NBK22183.
- 1 2 Clancy S (2008). "Genetic Mutation". Nature Education. 1 (1): 187.
- ↑ http://nervous-system.emedtv.com/tay-sachs-disease/causes-of-tay-sachs.html
- ↑ Ruder K (28 May 2004). "How Gene Mutations Cause Colorblindness". Genome News Network.
- ↑ Clutterbuck AJ (2011). "Genomic evidence of repeat-induced point mutation (RIP) in filamentous ascomycetes.". Fungal Genet Biol. 48 (3): 306–26. doi:10.1016/j.fgb.2010.09.002. PMID 20854921.
- 1 2 3 Selker EU, Cambareri EB, Jensen BC, Haack KR (December 1987). "Rearrangement of duplicated DNA in specialized cells of Neurospora". Cell. 51 (5): 741–752. doi:10.1016/0092-8674(87)90097-3. PMID 2960455.
- ↑ Graïa F, Lespinet O, Rimbault B, Dequard-Chablat M, Coppin E, Picard M (May 2001). "Genome quality control: RIP (repeat-induced point mutation) comes to Podospora". Mol Microbiol. 40 (3): 586–595. doi:10.1046/j.1365-2958.2001.02367.x. PMID 11359565.
- ↑ Ikeda K, Nakayashiki H, Kataoka T, Tamba H, Hashimoto Y, Tosa Y, Mayama S (September 2002). "Repeat-induced point mutation (RIP) in Magnaporthe grisea: implications for its sexual cycle in the natural field context". Mol Microbiol. 45 (5): 1355–1364. doi:10.1046/j.1365-2958.2002.03101.x. PMID 12207702.
- ↑ Idnurm A, Howlett BJ (June 2003). "Analysis of loss of pathogenicity mutants reveals that repeat-induced point mutations can occur in the Dothideomycete Leptosphaeria maculans". Fungal Genet Biol. 39 (1): 31–37. doi:10.1016/S1087-1845(02)00588-1. PMID 12742061.
- ↑ Cuomo CA, Güldener U, Xu JR, Trail F, Turgeon BG, Di Pietro A, Walton JD, Ma LJ, et al. (September 2007). "The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization". Science. 317 (5843): 1400–2. doi:10.1126/science.1143708. PMID 17823352.
- ↑ Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, Wasmann CC, Grimwood J, Schmutz J, et al. (August 2009). "The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion". PLoS Genet. 5 (8): e1000618. doi:10.1371/journal.pgen.1000618. PMC 2725324. PMID 19714214.
- ↑ Van de Wouw AP, Cozijnsen AJ, Hane JK, et al. (2010). "Evolution of linked avirulence effectors in Leptosphaeria maculans is affected by genomic environment and exposure to resistance genes in host plants". PLoS Pathog. 6 (11): e1001180. doi:10.1371/journal.ppat.1001180. PMC 2973834. PMID 21079787.
- ↑ Rep M, Kistler HC (August 2010). "The genomic organization of plant pathogenicity in Fusarium species". Curr. Opin. Plant Biol. 13 (4): 420–6. doi:10.1016/j.pbi.2010.04.004. PMID 20471307.
- ↑ Farman ML (August 2007). "Telomeres in the rice blast fungus Magnaporthe oryzae: the world of the end as we know it". FEMS Microbiol. Lett. 273 (2): 125–32. doi:10.1111/j.1574-6968.2007.00812.x. PMID 17610516.
- ↑ Fudal I, Ross S, Brun H, et al. (August 2009). "Repeat-induced point mutation (RIP) as an alternative mechanism of evolution toward virulence in Leptosphaeria maculans". Mol. Plant Microbe Interact. 22 (8): 932–41. doi:10.1094/MPMI-22-8-0932. PMID 19589069.
- ↑ Irelan JT, Hagemann AT, Selker EU (December 1994). "High frequency repeat-induced point mutation (RIP) is not associated with efficient recombination in Neurospora". Genetics. 138 (4): 1093–103. PMC 1206250. PMID 7896093.
- ↑ Freitag M, Williams RL, Kothe GO, Selker EU (2002). "A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa". Proc Natl Acad Sci U S A. 99 (13): 8802–7. doi:10.1073/pnas.132212899. PMC 124379. PMID 12072568.
- ↑ Lee DW, Freitag M, Selker EU, Aramayo R (2008). "A cytosine methyltransferase homologue is essential for sexual development in Aspergillus nidulans.". PLoS ONE. 3 (6): e2531. doi:10.1371/journal.pone.0002531. PMC 2432034. PMID 18575630.
- ↑ Malagnac F, Wendel B, Goyon C, Faugeron G, Zickler D, Rossignol JL, et al. (1997). "A gene essential for de novo methylation and development in Ascobolus reveals a novel type of eukaryotic DNA methyltransferase structure.". Cell. 91 (2): 281–90. doi:10.1016/S0092-8674(00)80410-9. PMID 9346245.
- ↑ Selker EU (1990). "Premeiotic instability of repeated sequences in Neurospora crassa.". Annu Rev Genet. 24: 579–613. doi:10.1146/annurev.ge.24.120190.003051. PMID 2150906.
- ↑ Barbieri, Marcello (2003). "The problem of generation". The organic codes: an introduction to semantic biology. Cambridge University Press. p. 13. ISBN 0-521-53100-4.
- ↑ Paweletz N (January 2001). "Walther Flemming: pioneer of mitosis research". Nat. Rev. Mol. Cell Biol. 2 (1): 72–5. doi:10.1038/35048077. PMID 11413469.
- ↑ Holmes, Frederic Lawrence (2001). Meselson, Stahl, and the replication of DNA : a history of "the most beautiful experiment in biology" (PDF). Yale University Press. ISBN 0-300-08540-0.
External links
| Wikimedia Commons has media related to Point mutation. |
- Point Mutation at the US National Library of Medicine Medical Subject Headings (MeSH)