Network motif
| Network science | ||||
|---|---|---|---|---|
| Network types | ||||
| Graphs | ||||
|
||||
| Models | ||||
|
||||
| ||||
|
||||
All networks, including biological networks, social networks, technological networks (e.g., computer networks and electrical circuits) and more, can be represented as graphs, which include a wide variety of subgraphs. One important local property of networks are so-called network motifs, which are defined as recurrent and statistically significant sub-graphs or patterns.
Network motifs are sub-graphs that repeat themselves in a specific network or even among various networks. Each of these sub-graphs, defined by a particular pattern of interactions between vertices, may reflect a framework in which particular functions are achieved efficiently. Indeed, motifs are of notable importance largely because they may reflect functional properties. They have recently gathered much attention as a useful concept to uncover structural design principles of complex networks.[1] Although network motifs may provide a deep insight into the network’s functional abilities, their detection is computationally challenging.
Definition
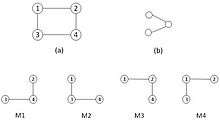
Let G = (V, E) and G′ = (V′, E′) be two graphs. Graph G′ is a sub-graph of graph G (written as G′ ⊆ G) if V′ ⊆ V and E′ ⊆ E ∩ (V′ × V′). If G′ ⊆ G and G′ contains all of the edges ‹u, v› ∈ E with u, v ∈ V′, then G′ is an induced sub-graph of G. We call G′ and G isomorphic (written as G′ ↔ G), if there exists a bijection (one-to-one) f:V′ → V with ‹u, v› ∈ E′ ⇔ ‹f(u), f(v)› ∈ E for all u, v ∈ V′. The mapping f is called an isomorphism between G and G′.[2]
When G″ ⊂ G and there exists an isomorphism between the sub-graph G″ and a graph G′, this mapping represents an appearance of G′ in G. The number of appearances of graph G′ in G is called the frequency FG of G′ in G. A graph is called recurrent (or frequent) in G, when its frequency FG(G′) is above a predefined threshold or cut-off value. We used terms pattern and frequent sub-graph in this review interchangeably. There is an ensemble Ω(G) of random graphs corresponding to the null-model associated to G. We should choose N random graphs uniformly from Ω(G) and calculate the frequency for a particular frequent sub-graph G′ in G. If the frequency of G′ in G is higher than its arithmetic mean frequency in N random graphs Ri, where 1 ≤ i ≤ N, we call this recurrent pattern significant and hence treat G′ as a network motif for G. For a small graph G′, the network G and a set of randomized networks R(G) ⊆ Ω(R), where , the Z-Score that has been defined by the following formula:

where μR(G′) and σR(G′) stand for mean and standard deviation frequency in set R(G), respectively.[3][4][5][6][7][8] The larger the Z(G′), the more significant is the sub-graph G′ as a motif. Alternatively, another measurement in statistical hypothesis testing that can be considered in motif detection is the P-Value, given as the probability of FR(G′) ≥ FG(G′) (as its null-hypothesis), where FR(G′) indicates the frequency of G' in a randomized network.[6] A sub-graph with P-value less than a threshold (commonly 0.01 or 0.05) will be treated as a significant pattern. The P-value is defined as

Where N indicates number of randomized networks, i is defined over an ensemble of randomized networks and the Kronecker delta function δ(c(i)) is one if the condition c(i) holds. The concentration [9][10] of a particular n-size sub-graph G′ in network G refers to the ratio of the sub-graph appearance in the network to the total n-size non-isomorphic sub-graphs’ frequencies, which is formulated by

where index i is defined over the set of all non-isomorphic n-size graphs. Another statistical measurement is defined for evaluating network motifs, but it is rarely used in known algorithms. This measurement is introduced by Picard et al. in 2008 and used the Poisson distribution, rather than the Gaussian normal distribution that is implicitly being used above.[11]
In addition, three specific concepts of sub-graph frequency have been proposed.[12] As figure illustrates, the first frequency concept F1 considers all matches of a graph in original network. This definition is similar to what we have introduced above. The second concept F2 is defined as the maximum number of edge-disjoint instances of a given graph in original network. And finally, the frequency concept F3 entails matches with disjoint edges and nodes. Therefore, the two concepts F2 and F3 restrict the usage of elements of the graph, and as can be inferred, the frequency of a sub-graph declines by imposing restrictions on network element usage. As a result, a network motif detection algorithm would pass over more candidate sub-graphs if we insist on frequency concepts F2 and F3.
History
This idea was first presented in 2002 by Uri Alon and his group [13] when network motifs were discovered in the gene regulation (transcription) network of the bacteria E. coli and then in a large set of natural networks. Since then, a considerable number of studies have been conducted on the subject. Some of these studies focus on the biological applications, while others focus on the computational theory of network motifs.
The biological studies endeavor to interpret the motifs detected for biological networks. For example, in work following,[13] the network motifs found in E. coli were discovered in the transcription networks of other bacteria[14] as well as yeast[15][16] and higher organisms.[17][18][19] A distinct set of network motifs were identified in other types of biological networks such as neuronal networks and protein interaction networks.[5][20][21]
The computational research has focused on improving existing motif detection tools to assist the biological investigations and allow larger networks to be analyzed. Several different algorithms have been provided so far, which are elaborated in the next section in chronological order.
Most recently, the acc-MOTIF tool to detect network motifs was released.[22]
Motif Discovery Algorithms
Various solutions have been proposed for the challenging problem of motif discovery. These algorithms can be classified under various paradigms such as exact counting methods, sampling methods, pattern growth methods and so on. However, motif discovery problem comprises two main steps: first, calculating the number of occurrences of a sub-graph and then, evaluating the sub-graph significance. The recurrence is significant if it is detectably far more than expected. Roughly speaking, the expected number of appearances of a sub-graph can be determined by a Null-model, which is defined by an ensemble of random networks with some of the same properties as the original network.
Here, a review on computational aspects of major algorithms is given and their related benefits and drawbacks from an algorithmic perspective are discussed.
mfinder
mfinder, the first motif-mining tool, implements two kinds of motif finding algorithms: a full enumeration and a sampling method. Until 2004, the only exact counting method for NM (network motif) detection was the brute-force one proposed by Milo et al..[3] This algorithm was successful for discovering small motifs, but using this method for finding even size 5 or 6 motifs was not computationally feasible. Hence, a new approach to this problem was needed.
Kashtan et al. [9] presented the first sampling NM discovery algorithm, which was based on edge sampling throughout the network. This algorithm estimates concentrations of induced sub-graphs and can be utilized for motif discovery in directed or undirected networks. The sampling procedure of the algorithm starts from an arbitrary edge of the network that leads to a sub-graph of size two, and then expands the sub-graph by choosing a random edge that is incident to the current sub-graph. After that, it continues choosing random neighboring edges until a sub-graph of size n is obtained. Finally, the sampled sub-graph is expanded to include all of the edges that exist in the network between these n nodes. When an algorithm uses a sampling approach, taking unbiased samples is the most important issue that the algorithm might address. The sampling procedure, however, does not take samples uniformly and therefore Kashtan et al. proposed a weighting scheme that assigns different weights to the different sub-graphs within network.[9] The underlying principle of weight allocation is exploiting the information of the sampling probability for each sub-graph, i.e. the probable sub-graphs will obtain comparatively less weights in comparison to the improbable sub-graphs; hence, the algorithm must calculate the sampling probability of each sub-graph that has been sampled. This weighting technique assists mfinder to determine sub-graph concentrations impartially.
In expanded to include sharp contrast to exhaustive search, the computational time of the algorithm surprisingly is asymptotically independent of the network size. An analysis of the computational time of the algorithm has shown that it takes O(nn) for each sample of a sub-graph of size n from the network. On the other hand, there is no analysis in [9] on the classification time of sampled sub-graphs that requires solving the graph isomorphism problem for each sub-graph sample. Additionally, an extra computational effort is imposed on the algorithm by the sub-graph weight calculation. But it is unavoidable to say that the algorithm may sample the same sub-graph multiple times – spending time without gathering any information.[10] In conclusion, by taking the advantages of sampling, the algorithm performs more efficiently than an exhaustive search algorithm; however, it only determines sub-graphs concentrations approximately. This algorithm can find motifs up to size 6 because of its main implementation, and as result it gives the most significant motif, not all the others too. Also, it is necessary to mention that this tool has no option of visual presentation. The sampling algorithm is shown briefly:
| mfinder |
|---|
| Definitions: Esis the set of picked edges. Vs is the set of all nodes that are touched by the edges in E. |
| Init Vs and Es to be empty sets.
1. Pick a random edge e1 = (vi, vj). Update Es = {e1}, Vs = {vi, vj} 2. Make a list L of all neighbor edges of Es. Omit from L all edges between members of Vs. 3. Pick a random edge e = {vk,vl} from L. Update Es = Es ⋃ {e}, Vs = Vs ⋃ {vk, vl}. 4. Repeat steps 2-3 until completing an n-node subgraph (until |Vs| = n). 5. Calculate the probability to sample the picked n-node subgraph. |
FPF (Mavisto)
Schreiber and Schwöbbermeyer [12] proposed an algorithm named flexible pattern finder (FPF) for extracting frequent sub-graphs of an input network and implemented it in a system named Mavisto.[23] Their algorithm exploits the downward closure property which is applicable for frequency concepts F2 and F3. The downward closure property asserts that the frequency for sub-graphs decrease monotonically by increasing the size of sub-graphs; however, this property does not hold necessarily for frequency concept F1. FPF is based on a pattern tree (see figure) consisting of nodes that represents different graphs (or patterns), where the parent of each node is a sub-graph of its children nodes; in other words, the corresponding graph of each pattern tree’s node is expanded by adding a new edge to the graph of its parent node.
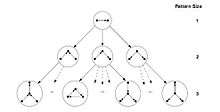
At first, the FPF algorithm enumerates and maintains the information of all matches of a sub-graph located at the root of the pattern tree. Then, one-by-one it builds child nodes of the previous node in the pattern tree by adding one edge supported by a matching edge in the target graph, and tries to expand all of the previous information about matches to the new sub-graph (child node). In next step, it decides whether the frequency of the current pattern is lower than a predefined threshold or not. If it is lower and if downward closure holds, FPF can abandon that path and not traverse further in this part of the tree; as a result, unnecessary computation is avoided. This procedure is continued until there is no remaining path to traverse.
The advantage of the algorithm is that it does not consider infrequent sub-graphs and tries to finish the enumeration process as soon as possible; therefore, it only spends time for promising nodes in the pattern tree and discards all other nodes. As an added bonus, the pattern tree notion permits FPF to be implemented and executed in a parallel manner since it is possible to traverse each path of the pattern tree independently. However, FPF is most useful for frequency concepts F2 and F3, because downward closure is not applicable to F1. Nevertheless, the pattern tree is still practical for F1 if the algorithm runs in parallel. Another advantage of the algorithm is that the implementation of this algorithm has no limitation on motif size, which makes it more amenable to improvements. The pseudocode of FPF (Mavisto) is shown below:
| Mavisto |
|---|
| Data: Graph G, target pattern size t, frequency concept F
Result: Set R of patterns of size t with maximum frequency. |
| R ← φ, fmax ← 0
P ←start pattern p1 of size 1 Mp1 ←all matches of p1 in G While P ≠ φ do Pmax ←select all patterns from P with maximum size. P ← select pattern with maximum frequency from Pmax Ε = ExtensionLoop(G, p, Mp) Foreach pattern p ∈ E If F = F1 then f ← size(Mp) Else f ← Maximum Independent set(F, Mp) End If size(p) = t then If f = fmax then R ← R ⋃ {p} Else if f > fmax then R ← {p}; fmax ← f End Else If F = F1 or f ≥ fmax then P ← P ⋃ {p} End End End End |
ESU (FANMOD)
The sampling bias of Kashtan et al. [9] provided great impetus for designing better algorithms for the NM discovery problem. Although Kashtan et al. tried to settle this drawback by means of a weighting scheme, this method imposed an undesired overhead on the running time as well a more complicated implementation. This tool is one of the most useful ones, as it supports visual options and also is an efficient algorithm with respect to time. But, it has a limitation on motif size as it does not allow searching for motifs of size 9 or higher because of the way the tool is implemented.
Wernicke [10] introduced an algorithm named RAND-ESU that provides a significant improvement over mfinder.[9] This algorithm, which is based on the exact enumeration algorithm ESU, has been implemented as an application called FANMOD.[10] RAND-ESU is a NM discovery algorithm applicable for both directed and undirected networks, effectively exploits an unbiased node sampling throughout the network, and prevents overcounting sub-graphs more than once. Furthermore, RAND-ESU uses a novel analytical approach called DIRECT for determining sub-graph significance instead of using an ensemble of random networks as a Null-model. The DIRECT method estimates the sub-graph concentration without explicitly generating random networks.[10] Empirically, the DIRECT method is more efficient in comparison with the random network ensemble in case of sub-graphs with a very low concentration; however, the classical Null-model is faster than the DIRECT method for highly concentrated sub-graphs.[3][10] In the following, we detail the ESU algorithm and then we show how this exact algorithm can be modified efficiently to RAND-ESU that estimates sub-graphs concentrations.
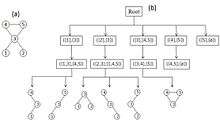
The algorithms ESU and RAND-ESU are fairly simple, and hence easy to implement. ESU first finds the set of all induced sub-graphs of size k, let Sk be this set. ESU can be implemented as a recursive function; the running of this function can be displayed as a tree-like structure of depth k, called the ESU-Tree (see figure). Each of the ESU-Tree nodes indicate the status of the recursive function that entails two consecutive sets SUB and EXT. SUB refers to nodes in the target network that are adjacent and establish a partial sub-graph of size |SUB| ≤ k. If |SUB| = k, the algorithm has found an induced complete sub-graph, so Sk = SUB ∪ Sk. However, if |SUB| < k, the algorithm must expand SUB to achieve cardinality k. This is done by the EXT set that contains all the nodes that satisfy two conditions: First, each of the nodes in EXT must be adjacent to at least one of the nodes in SUB; second, their numerical labels must be larger than the labels of SUB nodes. The first condition makes sure that the expansion of SUB nodes yields a connected graph and the second condition causes ESU-Tree leaves (see figure) to be distinct; as a result, it prevents overcounting. Note that, the EXT set is not a static set, so in each step it may expand by some new nodes that do not breach the two conditions. The next step of ESU involves classification of sub-graphs placed in the ESU-Tree leaves into non-isomorphic size-k graph classes; consequently, ESU determines sub-graphs frequencies and concentrations. This stage has been implemented simply by employing McKay’s nauty algorithm,[24][25] which classifies each sub-graph by performing a graph isomorphism test. Therefore, ESU finds the set of all induced k-size sub-graphs in a target graph by a recursive algorithm and then determines their frequency using an efficient tool.
How has the exact ESU been algorithm modified to RAND-ESU that estimates sub-graph concentrations? The procedure of implementing RAND-ESU is quite straightforward and is one of the main advantages of FANMOD. One can change the ESU algorithm to explore just a portion of the ESU-Tree leaves by applying a probability value 0 ≤ pd ≤ 1 for each level of the ESU-Tree and oblige ESU to traverse each child node of a node in level d-1 with probability pd. This new algorithm is called RAND-ESU. Evidently, when pd = 1 for all levels, RAND-ESU acts like ESU. For pd = 0 the algorithm finds nothing. Note that, this procedure ensures that the chances of visiting each leaf of the ESU-Tree are the same, resulting in unbiased sampling of sub-graphs through the network. The probability of visiting each leaf is ∏dpd and this is identical for all of the ESU-Tree leaves; therefore, this method guarantees unbiased sampling of sub-graphs from the network. Nonetheless, determining the value of pd for 1 ≤ d ≤ k is another issue that must be determined manually by an expert to get precise results of sub-graph concentrations.[8] While there is no lucid prescript for this matter, the Wernicke provides some general observations that may help in determining p_d values. In summary, RAND-ESU is a very fast algorithm for NM discovery in the case of induced sub-graphs supporting unbiased sampling method. Although, the main ESU algorithm and so the FANMOD tool is known for discovering induced sub-graphs, there is trivial modification to ESU which makes it possible for finding non-induced sub-graphs, too. The pseudo code of ESU (FANMOD) is shown below:
| Enumeration of ESU (FANMOD) |
|---|
| EnumerateSubgraphs(G,k)
Input: A graph G = (V, E) and an integer 1 ≤ k ≤ |V|. Output: All size-k subgraphs in G. for each vertex v ∈ V do VExtension ← {u ∈ N({v}) | u > v} call ExtendSubgraph({v}, VExtension, v) endfor |
| ExtendSubgraph(VSubgraph, VExtension, v)
if |VSubgraph| = k then output G[VSubgraph] and return while VExtension ≠ ∅ do Remove an arbitrarily chosen vertex w from VExtension VExtension′ ← VExtension ∪ {u ∈ Nexcl(w, VSubgraph) | u > v} call ExtendSubgraph(VSubgraph ∪ {w}, VExtension′, v) return |
NeMoFinder
Chen et al. [26] introduced a new NM discovery algorithm called NeMoFinder, which adapts the idea in SPIN [27] to extract frequent trees and after that expands them into non-isomorphic graphs.[8] NeMoFinder utilizes frequent size-n trees to partition the input network into a collection of size-n graphs, afterward finding frequent size-n sub-graphs by expansion of frequent trees edge-by-edge until getting a complete size-n graph Kn. The algorithm finds NMs in undirected networks and is not limited to extracting only induced sub-graphs. Furthermore, NeMoFinder is an exact enumeration algorithm and is not based on a sampling method. As Chen et al. claim, NeMoFinder is applicable for detecting relatively large NMs, for instance, finding NMs up to size-12 from the whole S. cerevisiae (yeast) PPI network as the authors claimed.[28]
NeMoFinder consists of three main steps. First, finding frequent size-n trees, then utilizing repeated size-n trees to divide the entire network into a collection of size-n graphs, finally, performing sub-graph join operations to find frequent size-n sub-graphs.[26] In the first step, the algorithm detects all non-isomorphic size-n trees and mappings from a tree to the network. In the second step, the ranges of these mappings are employed to partition the network into size-n graphs. Up to this step, there is no distinction between NeMoFinder and an exact enumeration method. However, a large portion of non-isomorphic size-n graphs still remain. NeMoFinder exploits a heuristic to enumerate non-tree size-n graphs by the obtained information from the preceding steps. The main advantage of the algorithm is in the third step, which generates candidate sub-graphs from previously enumerated sub-graphs. This generation of new size-n sub-graphs is done by joining each previous sub-graph with derivative sub-graphs from itself called cousin sub-graphs. These new sub-graphs contain one additional edge in comparison to the previous sub-graphs. However, there exist some problems in generating new sub-graphs: There is no clear method to derive cousins from a graph, joining a sub-graph with its cousins leads to redundancy in generating particular sub-graph more than once, and cousin determination is done by a canonical representation of the adjacency matrix which is not closed under join operation. NeMoFinder is an efficient network motif finding algorithm for motifs up to size 12 only for protein-protein interaction networks, which are presented as undirected graphs. And it is not able to work on directed networks which are so important in the field of complex and biological networks. The pseudocode of NeMoFinder is shown below:
| NeMoFinder |
|---|
| Input:
G - PPI network; N - Number of randomized networks; K - Maximal network motif size; F - Frequency threshold; S - Uniqueness threshold; Output: U - Repeated and unique network motif set; D ← ∅; for motif-size k from 3 to K do T ← FindRepeatedTrees(k); GDk ← GraphPartition(G, T) D ← D ∪ T; D′ ← T; i ← k; while D″ = ∅ and i ≤ k × (k - 1) / 2 do D′ ← FindRepeatedGraphs(k, i, D′); D ← D ∪ D′; i ← i + 1; end while end for for counter i from 1 to N do Grand ← RandomizedNetworkGeneration(); for each g ∈ D do GetRandFrequency(g, Grand); end for end for U ← ∅; for each g ∈ D do s ← GetUniqunessValue(g); if s ≥ S then U ← U ∪ {g}; end if end for return U |
Grochow-Kellis
Grochow and Kellis [29] proposed an exact algorithm for enumerating sub-graph appearances. The algorithm is based on a motif-centric approach, which means that the frequency of a given sub-graph,called the query graph, is exhaustively determined by searching for all possible mappings from the query graph into the larger network. It is claimed [29] that a motif-centric method in comparison to network-centric methods has some beneficial features. First of all it avoids the increased complexity of sub-graph enumeration. Also, by using mapping instead of enumerating, it enables an improvement in the isomorphism test. To improve the performance of the algorithm, since it is an inefficient exact enumeration algorithm, the authors introduced a fast method which is called symmetry-breaking conditions. During straightforward sub-graph isomorphism tests, a sub-graph may be mapped to the same sub-graph of the query graph multiple times. In the Grochow-Kellis (GK) algorithm symmetry-breaking is used to avoid such multiple mappings. Here we introduce the GK algorithm and the symmetry-breaking condition which eliminates redundant isomorphism tests.
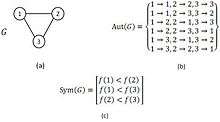
The GK algorithm discovers the whole set of mappings of a given query graph to the network in two major steps. It starts with the computation of symmetry-breaking conditions of the query graph. Next, by means of a branch-and-bound method, the algorithm tries to find every possible mapping from the query graph to the network that meets the associated symmetry-breaking conditions. An example of the usage of symmetry-breaking conditions in GK algorithm is demonstrated in figure.
As it is mentioned above, the symmetry-breaking technique is a simple mechanism that precludes spending time finding a sub-graph more than once due to its symmetries.[29][30] Note that, computing symmetry-breaking conditions requires finding all automorphisms of a given query graph. Even though, there is no efficient (or polynomial time) algorithm for the graph automorphism problem, this problem can be tackled efficiently in practice by McKay’s tools.[24][25] As it is claimed, using symmetry-breaking conditions in NM detection lead to save a great deal of running time. Moreover, it can be inferred from the results in [29][30] that using the symmetry-breaking conditions results in high efficiency particularly for directed networks in comparison to undirected networks. The symmetry-breaking conditions used in the GK algorithm are similar to the restriction which ESU algorithm applies to the labels in EXT and SUB sets. In conclusion, the GK algorithm computes the exact number of appearance of a given query graph in a large complex network and exploiting symmetry-breaking conditions improves the algorithm performance. Also, GK algorithm is one of the known algorithms having no limitation for motif size in implementation and potentially it can find motifs of any size.
Color-Coding Approach
Most algorithms in the field of NM discovery are used to find induced sub-graphs of a network. In 2008, Noga Alon et al. [31] introduced an approach for finding non-induced sub-graphs too. Their technique works on undirected networks such as PPI ones. Also, it counts non-induced trees and bounded treewidth sub-graphs. This method is applied for sub-graphs of size up to 10.
This algorithm counts the number of non-induced occurrences of a tree T with k = O(logn) vertices in a network G with n vertices as follows:
1. Color coding. Color each vertex of input network G independently and uniformly at random with one of the k colors.
2. Counting. Apply a dynamic programming routine to count the number of non-induced occurrences of T in which each vertex has a unique color. For more details on this step, see.[31]
3. Repeat the above two steps O(ek) times and add up the number of occurrences of T to get an estimate on the number of its occurrences in G.
As available PPI networks are far from complete and error free, this approach is suitable for NM discovery for such networks. As Grochow-Kellis Algorithm and this one are the ones popular for non-induced sub-graphs, it is worth to mention that the algorithm introduced by Alon et al. is less time consuming than the Grochow-Kellis Algorithm.[31]
MODA
Omidi et al. [32] introduced a new algorithm for motif detection named MODA which is applicable for induced and non-induced NM discovery in undirected networks. It is based on the motif-centric approach discussed in the Grochow-Kellis algorithm section. It is very important to distinguish motif-centric algorithms such as MODA and GK algorithm because of their ability to work as query-finding algorithms. This feature allows such algorithms to be able to find a single motif query or a small number of motif queries (not all possible sub-graphs of a given size) with larger sizes. As the number of possible non-isomorphic sub-graphs increases exponentially with sub-graph size, for large size motifs (even larger than 10), the network-centric algorithms, those looking for all possible sub-graphs, face a problem. Although motif-centric algorithms also have problems in discovering all possible large size sub-graphs, but their ability to find small numbers of them is sometimes a significant property.
Using a hierarchical structure called an expansion tree, the MODA algorithm is able to extract NMs of a given size systematically and similar to FPF that avoids enumerating unpromising sub-graphs; MODA takes into consideration potential queries (or candidate sub-graphs) that would result in frequent sub-graphs. Despite the fact that MODA resembles FPF in using a tree like structure, the expansion tree is applicable merely for computing frequency concept F1. As we will discuss next, the advantage of this algorithm is that it does not carry out the sub-graph isomorphism test for non-tree query graphs. Additionally, it utilizes a sampling method in order to speed up the running time of the algorithm.
Here is the main idea: by a simple criterion one can generalize a mapping of a k-size graph into the network to its same size supergraphs. For example, suppose there is mapping f(G) of graph G with k nodes into the network and we have a same size graph G′ with one more edge ‹u, v›; fG will map G′ into the network, if there is an edge ‹fG(u), fG(v)› in the network. As a result, we can exploit the mapping set of a graph to determine the frequencies of its same order supergraphs simply in O(1) time without carrying out sub-graph isomorphism testing. The algorithm starts ingeniously with minimally connected query graphs of size k and finds their mappings in the network via sub-graph isomorphism. After that, with conservation of the graph size, it expands previously considered query graphs edge-by-edge and computes the frequency of these expanded graphs as mentioned above. The expansion process continues until reaching a complete graph Kk (fully connected with k(k-1)⁄2 edge).
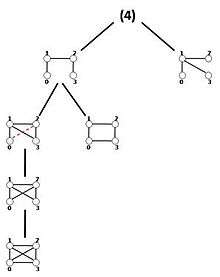
As discussed above, the algorithm starts by computing sub-tree frequencies in the network and then expands sub-trees edge by edge. One way to implement this idea is called the expansion tree Tk for each k. Figure shows the expansion tree for size-4 sub-graphs. Tk organizes the running process and provides query graphs in a hierarchical manner. Strictly speaking, the expansion tree Tk is simply a directed acyclic graph or DAG, with its root number k indicating the graph size existing in the expansion tree and each of its other nodes containing the adjacency matrix of a distinct k-size query graph. Nodes in the first level of Tk are all distinct k-size trees and by traversing Tk in depth query graphs expand with one edge at each level. A query graph in a node is a sub-graph of the query graph in a node’s child with one edge difference. The longest path in Tk consists of (k2-3k+4)/2 edges and is the path from the root to the leaf node holding the complete graph. Generating expansion trees can be done by a simple routine which is explained in.[32]
MODA traverses Tk and when it extracts query trees from the first level of Tk it computes their mapping sets and saves these mappings for the next step. For non-tree queries from Tk, the algorithm extracts the mappings associated with the parent node in Tk and determines which of these mappings can support the current query graphs. The process will continue until the algorithm gets the complete query graph. The query tree mappings are extracted using the Grochow-Kellis algorithm. For computing the frequency of non-tree query graphs, the algorithm employs a simple routine that takes O(1) steps. In addition, MODA exploits a sampling method where the sampling of each node in the network is linearly proportional to the node degree, the probability distribution is exactly similar to the well-known Barabási-Albert preferential attachment model in the field of complex networks.[33] This approach generates approximations; however, the results are almost stable in different executions since sub-graphs aggregate around highly connected nodes.[34] The pseudocode of MODA is shown below:
| MODA |
|---|
| Input: G: Input graph, k: sub-graph size, Δ: threshold value
Output: Frequent Subgraph List: List of all frequent k-size sub-graphs Note: FG: set of mappings from G in the input graph G fetch Tk do G′ = Get-Next-BFS(Tk) // G′ is a query graph if |E(G′)| = (k – 1) call Mapping-Module(G′, G) else call Enumerating-Module(G′, G, Tk) end if save F2 if |FG| > Δ then add G′ into Frequent Subgraph List end if Until |E(G')| = (k – 1)/2) return Frequent Subgraph List |
Kavosh
A recently introduced algorithm named Kavosh [35] aims at improved main memory usage. Kavosh is usable to detect NM in both directed and undirected networks. The main idea of the enumeration is similar to the GK and MODA algorithms, which first find all k-size sub-graphs that a particular node participated in, then remove the node, and subsequently repeat this process for the remaining nodes.[35]
For counting the sub-graphs of size k that include a particular node, trees with maximum depth of k, rooted at this node and based on neighborhood relationship are implicitly built. Children of each node include both incoming and outgoing adjacent nodes. To descend the tree, a child is chosen at each level with the restriction that a particular child can be included only if it has not been included at any upper level. After having descended to the lowest level possible, the tree is again ascended and the process is repeated with the stipulation that nodes visited in earlier paths of a descendent are now considered unvisited nodes. A final restriction in building trees is that all children in a particular tree must have numerical labels larger than the label of the root of the tree. The restrictions on the labels of the children are similar to the conditions which GK and ESU algorithm use to avoid overcounting sub-graphs.
The protocol for extracting sub-graphs makes use of the compositions of an integer. For the extraction of sub-graphs of size k, all possible compositions of the integer k-1 must be considered. The compositions of k-1 consist of all possible manners of expressing k-1 as a sum of positive integers. Summations in which the order of the summands differs are considered distinct. A composition can be expressed as k2,k3,…,km where k2 + k3 + … + km = k-1. To count sub-graphs based on the composition, ki nodes are selected from the i-th level of the tree to be nodes of the sub-graphs (i = 2,3,…,m). The k-1 selected nodes along with the node at the root define a sub-graph within the network. After discovering a sub-graph involved as a match in the target network, in order to be able to evaluate the size of each class according to the target network, Kavosh employs the nauty algorithm [24][25] in the same way as FANMOD. The enumeration part of Kavosh algorithm is shown below:
| Enumeration of Kavosh |
|---|
| Enumerate_Vertex(G, u, S, Remainder, i)
Input: G: Input graph if Remainder = 0 then |
| Validate(G, Parents, u) Input: G: input graph, Parents: selected vertices of last layer, u: Root vertex. ValList ← NILL |
Recently a Cytoscape plugin called CytoKavosh [36] is developed for this software. It is available via Cytoscape web page .
G-Tries
In 2010, Pedro Ribeiro and Fernando Silva proposed a novel data structure for storing a collection of sub-graphs, called a g-trie.[37] This data structure, which is conceptually akin to a prefix tree, stores sub-graphs according to their structures and finds occurrences of each of these sub-graphs in a larger graph. One of the noticeable aspects of this data structure is that coming to the network motif discovery, the sub-graphs in the main network are needed to be evaluated. So, there is no need to find the ones in random network which are not in the main network. This can be one of the time-consuming parts in the algorithms in which all sub-graphs in random networks are derived.
A g-trie is a multiway tree that can store a collection of graphs. Each tree node contains information about a single graph vertex and its corresponding edges to ancestor nodes. A path from the root to a leaf corresponds to one single graph. Descendants of a g-trie node share a common sub-graph. Constructing a g-trie is well described in.[37] After constructing a g-trie, the counting part takes place. The main idea in counting process is to backtrack by all possible sub-graphs, but at the same time do the isomorphism tests. This backtracking technique is essentially the same technique employed by other motif-centric approaches like MODA and GK algorithms. Taking advantage of common substructures in the sense that at a given time there is a partial isomorphic match for several different candidate sub-graphs.
Among the mentioned algorithms, G-Tries is the fastest. But, the excessive use of memory is the drawback of this algorithm, which might limit the size of discoverable motifs by a personal computer with average memory.
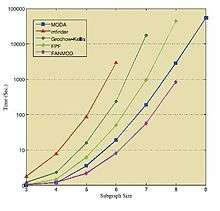
Comparison
Tables and figure below show the results of running the mentioned algorithms on different standard networks. These results are taken from the corresponding sources,[32][35][37] thus they should be treated individually.
| Network | Size | Census Original Network | Average Census on Random Networks | ||||
|---|---|---|---|---|---|---|---|
| FANMOD | GK | G-Trie | FANMOD | GK | G-Trie | ||
| Dolphins | 5 | 0.07 | 0.03 | 0.01 | 0.13 | 0.04 | 0.01 |
| 6 | 0.48 | 0.28 | 0.04 | 1.14 | 0.35 | 0.07 | |
| 7 | 3.02 | 3.44 | 0.23 | 8.34 | 3.55 | 0.46 | |
| 8 | 19.44 | 73.16 | 1.69 | 67.94 | 37.31 | 4.03 | |
| 9 | 100.86 | 2984.22 | 6.98 | 493.98 | 366.79 | 24.84 | |
| Circuit | 6 | 0.49 | 0.41 | 0.03 | 0.55 | 0.24 | 0.03 |
| 7 | 3.28 | 3.73 | 0.22 | 3.53 | 1.34 | 0.17 | |
| 8 | 17.78 | 48.00 | 1.52 | 21.42 | 7.91 | 1.06 | |
| Social | 3 | 0.31 | 0.11 | 0.02 | 0.35 | 0.11 | 0.02 |
| 4 | 7.78 | 1.37 | 0.56 | 13.27 | 1.86 | 0.57 | |
| 5 | 208.30 | 31.85 | 14.88 | 531.65 | 62.66 | 22.11 | |
| Yeast | 3 | 0.47 | 0.33 | 0.02 | 0.57 | 0.35 | 0.02 |
| 4 | 10.07 | 2.04 | 0.36 | 12.90 | 2.25 | 0.41 | |
| 5 | 268.51 | 34.10 | 12.73 | 400.13 | 47.16 | 14.98 | |
| Power | 3 | 0.51 | 1.46 | 0.00 | 0.91 | 1.37 | 0.01 |
| 4 | 1.38 | 4.34 | 0.02 | 3.01 | 4.40 | 0.03 | |
| 5 | 4.68 | 16.95 | 0.10 | 12.38 | 17.54 | 0.14 | |
| 6 | 20.36 | 95.58 | 0.55 | 67.65 | 92.74 | 0.88 | |
| 7 | 101.04 | 765.91 | 3.36 | 408.15 | 630.65 | 5.17 | |
| Size → | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
|---|---|---|---|---|---|---|---|---|---|
| Networks ↓ | Algorithms ↓ | ||||||||
| E. coli | Kavosh | 0.30 | 1.84 | 14.91 | 141.98 | 1374.0 | 13173.7 | 121110.3 | 1120560.1 |
| FANMOD | 0.81 | 2.53 | 15.71 | 132.24 | 1205.9 | 9256.6 | - | - | |
| Mavisto | 13532 | - | - | - | - | - | - | - | |
| Mfinder | 31.0 | 297 | 23671 | - | - | - | - | - | |
| Electronic | Kavosh | 0.08 | 0.36 | 8.02 | 11.39 | 77.22 | 422.6 | 2823.7 | 18037.5 |
| FANMOD | 0.53 | 1.06 | 4.34 | 24.24 | 160 | 967.99 | - | - | |
| Mavisto | 210.0 | 1727 | - | - | - | - | - | - | |
| Mfinder | 7 | 14 | 109.8 | 2020.2 | - | - | - | - | |
| Social | Kavosh | 0.04 | 0.23 | 1.63 | 10.48 | 69.43 | 415.66 | 2594.19 | 14611.23 |
| FANMOD | 0.46 | 0.84 | 3.07 | 17.63 | 117.43 | 845.93 | - | - | |
| Mavisto | 393 | 1492 | - | - | - | - | - | - | |
| Mfinder | 12 | 49 | 798 | 181077 | - | - | - | - |
Classification of Algorithms
As seen in the table, motif discovery algorithms can be divided into two general categories: those based on exact counting and those using statistical sampling and estimations instead. Because the second group does not count all the occurrences of a subgraph in the main network, the algorithms belonging to this group are faster, but they might yield in biased and unrealistic results.
In the next level, the exact counting algorithms can be classified to network-centric and subgraph-centric methods. The algorithms of the first class search the given network for all subgraphs of a given size, while the algorithms falling into the second class first generate different possible non-isomorphic graphs of the given size, and then explore the network for each generated subgraph separately. Each approach has its advantages and disadvantages which are discussed above.
On the other hand, estimation methods might utilize color-coding approach as described before. Other approaches used in this category usually skip some subgraphs during enumeration (e.g., as in FANMOD) and base their estimation on the enumerated subgraphs.
Furthermore, table indicates whether an algorithm can be used for directed or undirected networks as well as induced or non-induced subgraphs. For more information refer to the provided web links or lab addresses.
| Counting Method | Basis | Name | Directed / Undirected | Induced / Non-Induced |
|---|---|---|---|---|
| Exact | Network-Centric | mfinder | Both | Induced |
| ESU (FANMOD) | Both | Induced | ||
| Kavosh (used in CytoKavosh) | Both | Induced | ||
| G-Tries | Both | Induced | ||
| PGD | Undirected | Induced | ||
| Subgraph-Centric | FPF (Mavisto) | Both | Induced | |
| NeMoFinder | Undirected | Induced | ||
| Grochow-Kellis | Both | Both | ||
| MODA | Both | Both | ||
| Estimation / Sampling | Color-Coding Approach | N. Alon et al.’s Algorithm | Undirected | Non-Induced |
| Other Approaches | mfinder | Both | Induced | |
| ESU (FANMOD) | Both | Induced |
| Algorithm | Lab / Dept. Name | Department / School | Institute | Address | |
|---|---|---|---|---|---|
| mfinder | Uri Alon's Group | Department of Molecular Cell Biology | Weizmann Institute of Science | Rehovot, Israel, Wolfson, Rm. 607 | urialon at weizmann.ac.il |
| FPF (Mavisto) | ---- | ---- | Leibniz-Institut für Pflanzengenetik und Kulturpflanzenforschung (IPK) | Corrensstraße 3, D-06466 Stadt Seeland, OT Gatersleben, Deutschland | schreibe at ipk-gatersleben.de |
| ESU (FANMOD) | Lehrstuhl Theoretische Informatik I | Institut für Informatik | Friedrich-Schiller-Universität Jena | Ernst-Abbe-Platz 2,D-07743 Jena, Deutschland | sebastian.wernicke at gmail.com |
| NeMoFinder | ---- | School of Computing | National University of Singapore | Singapore 119077 | chenjin at comp.nus.edu.sg |
| Grochow-Kellis | Theory Group | Computer Science | The University of Chicago | 1100 East 58th Street, Chicago, IL 60637 | joshuag at cs.uchicago.edu |
| N. Alon et al.’s Algorithm | Department of Pure Mathematics | School of Mathematical Sciences | Tel Aviv University | Tel Aviv 69978, Israel | nogaa at post.tau.ac.il |
| MODA | Laboratory of Systems Biology and Bioinformatics (LBB) | Institute of Biochemistry and Biophysics (IBB) | University of Tehran | Enghelab Square, Enghelab Ave, Tehran, Iran | amasoudin at ibb.ut.ac.ir |
| Kavosh (used in CytoKavosh) | Laboratory of Systems Biology and Bioinformatics (LBB) | Institute of Biochemistry and Biophysics (IBB) | University of Tehran | Enghelab Square, Enghelab Ave, Tehran, Iran | amasoudin at ibb.ut.ac.ir |
| G-Tries | Center for Research in Advanced Computing Systems | Computer Science | University of Porto | Rua Campo Alegre 1021/1055, Porto, Portugal | pribeiro at dcc.fc.up.pt |
| PGD | Network Learning and Discovery Lab | Department of Computer Science | Purdue University | Purdue University, 305 N University St, West Lafayette, IN 47907 | nkahmed@purdue.edu |
Well-Established Motifs and Their Functions
Much experimental work has been devoted to understanding network motifs in gene regulatory networks. These networks control which genes are expressed in the cell in response to biological signals. The network is defined such that genes are nodes, and directed edges represent the control of one gene by a transcription factor (regulatory protein that binds DNA) encoded by another gene. Thus, network motifs are patterns of genes regulating each other's transcription rate. When analyzing transcription networks, it is seen that the same network motifs appear again and again in diverse organisms from bacteria to human. The transcription network of E. coli and yeast, for example, is made of three main motif families, that make up almost the entire network. The leading hypothesis is that the network motif were independently selected by evolutionary processes in a converging manner,[38][39] since the creation or elimination of regulatory interactions is fast on evolutionary time scale, relative to the rate at which genes change,[38][39][40] Furthermore, experiments on the dynamics generated by network motifs in living cells indicate that they have characteristic dynamical functions. This suggests that the network motif serve as building blocks in gene regulatory networks that are beneficial to the organism.
The functions associated with common network motifs in transcription networks were explored and demonstrated by several research projects both theoretically and experimentally. Below are some of the most common network motifs and their associated function.
Negative auto-regulation (NAR)

One of simplest and most abundant network motifs in E. coli is negative auto-regulation in which a transcription factor (TF) represses its own transcription. This motif was shown to perform two important functions. The first function is response acceleration. NAR was shown to speed-up the response to signals both theoretically [41] and experimentally. This was first shown in a synthetic transcription network[42] and later on in the natural context in the SOS DNA repair system of E .coli.[43] The second function is increased stability of the auto-regulated gene product concentration against stochastic noise, thus reducing variations in protein levels between different cells.[44][45][46]
Positive auto-regulation (PAR)
Positive auto-regulation (PAR) occurs when a transcription factor enhances its own rate of production. Opposite to the NAR motif this motif slows the response time compared to simple regulation.[47] In the case of a strong PAR the motif may lead to a bimodal distribution of protein levels in cell populations.[48]
Feed-forward loops (FFL)
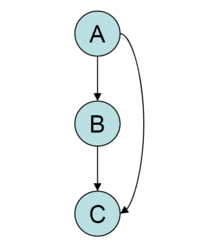
This motif is commonly found in many gene systems and organisms. The motif consists of three genes and three regulatory interactions. The target gene C is regulated by 2 TFs A and B and in addition TF B is also regulated by TF A . Since each of the regulatory interactions may either be positive or negative there are possibly eight types of FFL motifs.[49] Two of those eight types: the coherent type 1 FFL (C1-FFL) (where all interactions are positive) and the incoherent type 1 FFL (I1-FFL) (A activates C and also activates B which represses C) are found much more frequently in the transcription network of E. coli and yeast than the other six types.[49][50] In addition to the structure of the circuitry the way in which the signals from A and B are integrated by the C promoter should also be considered. In most of the cases the FFL is either an AND gate (A and B are required for C activation) or OR gate (either A or B are sufficient for C activation) but other input function are also possible.
Coherent type 1 FFL (C1-FFL)
The C1-FFL with an AND gate was shown to have a function of a ‘sign-sensitive delay’ element and a persistence detector both theoretically [49] and experimentally[51] with the arabinose system of E. coli. This means that this motif can provide pulse filtration in which short pulses of signal will not generate a response but persistent signals will generate a response after short delay. The shut off of the output when a persistent pulse is ended will be fast. The opposite behavior emerges in the case of a sum gate with fast response and delayed shut off as was demonstrated in the flagella system of E. coli.[52]
Incoherent type 1 FFL (I1-FFL)
The I1-FFL is a pulse generator and response accelerator. The two signal pathways of the I1-FFL act in opposite directions where one pathway activates Z and the other represses it. When the repression is complete this leads to a pulse-like dynamics. It was also demonstrated experimentally that the I1-FFL can serve as response accelerator in a way which is similar to the NAR motif. The difference is that the I1-FFL can speed-up the response of any gene and not necessarily a transcription factor gene.[53] An additional function was assigned to the I1-FFL network motif: it was shown both theoretically and experimentally that the I1-FFL can generate non-monotonic input function in both a synthetic [54] and native systems.[55] Finally, expression units that incorporate incoherent feedforward control of the gene product provide adaptation to the amount of DNA template and can be superior to simple combinations of constitutive promoters.[56] Feedforward regulation displayed better adaptation than negative feedback, and circuits based on RNA interference were the most robust to variation in DNA template amounts.[56]
Multi-output FFLs
In some cases the same regulators X and Y regulate several Z genes of the same system. By adjusting the strength of the interactions this motif was shown to determine the temporal order of gene activation. This was demonstrated experimentally in the flagella system of E. coli.[57]
Single-input modules (SIM)
This motif occurs when a single regulator regulates a set of genes with no additional regulation. This is useful when the genes are cooperatively carrying out a specific function and therefore always need to be activated in a synchronized manner. By adjusting the strength of the interactions it can create temporal expression program of the genes it regulates.[58]
In the literature, Multiple-input modules (MIM) arose as a generalization of SIM. However, the precise definitions of SIM and MIM have been a source of inconsistency. There are attempts to provide orthogonal definitions for canonical motifs in biological networks and algorithms to enumerate them, especially SIM, MIM and Bi-Fan (2x2 MIM).[59]
Dense overlapping regulons (DOR)
This motif occurs in the case that several regulators combinatorially control a set of genes with diverse regulatory combinations. This motif was found in E. coli in various systems such as carbon utilization, anaerobic growth, stress response and others.[13][18] In order to better understand the function of this motif one has to obtain more information about the way the multiple inputs are integrated by the genes. Kaplan et al.[60] has mapped the input functions of the sugar utilization genes in E. coli, showing diverse shapes.
Activity motifs
An interesting generalization of the network-motifs, activity motifs are over occurring patterns that can be found when nodes and edges in the network are annotated with quantitative features. For instance, when edges in a metabolic pathways are annotated with the magnitude or timing of the corresponding gene expression, some patterns are over occurring given the underlying network structure.[61]
Criticism
An assumption (sometimes more sometimes less implicit) behind the preservation of a topological sub-structure is that it is of a particular functional importance. This assumption has recently been questioned. Some authors have argued that motifs, like bi-fan motifs, might show a variety depending on the network context, and therefore,[62] structure of the motif does not necessarily determine function. Network structure certainly does not always indicate function; this is an idea that has been around for some time, for an example see the Sin operon.[63]
Most analyses of motif function are carried out looking at the motif operating in isolation. Recent research[64] provides good evidence that network context, i.e. the connections of the motif to the rest of the network, is too important to draw inferences on function from local structure only — the cited paper also reviews the criticisms and alternative explanations for the observed data. An analysis of the impact of a single motif module on the global dynamics of a network is studied in.[65] Yet another recent work suggests that certain topological features of biological networks naturally give rise to the common appearance of canonical motifs, thereby questioning whether frequencies of occurrences are reasonable evidence that the structures of motifs are selected for their functional contribution to the operation of networks.[66]
See also
References
- ↑ Masoudi-Nejad A, Schreiber F, Razaghi MK Z (2012). "Building Blocks of Biological Networks: A Review on Major Network Motif Discovery Algorithms". IET Systems Biology, in press.
- ↑ Diestel R (2005). "Graph Theory (Graduate Texts in Mathematics)". 173. New York: Springer-Verlag Heidelberg.
- 1 2 3 Milo R, Shen-Orr SS, Itzkovitz S, Kashtan N, Chklovskii D, Alon U (2002). "Network motifs: simple building blocks of complex networks". Science. 298 (5594): 824–827. Bibcode:2002Sci...298..824M. doi:10.1126/science.298.5594.824. PMID 12399590.
- ↑ Albert R, Barabási AL (2002). "Statistical mechanics of complex networks". Reviews of Modern Physics. 74: 47–49. Bibcode:2002RvMP...74...47A. doi:10.1103/RevModPhys.74.47.
- 1 2 Milo R, Itzkovitz S, Kashtan N, Levitt R, Shen-Orr S, Ayzenshtat I, Sheffer M, Alon U (2004). "Superfamilies of designed and evolved networks". Science. 303 (5663): 1538–1542. Bibcode:2004Sci...303.1538M. doi:10.1126/science.1089167. PMID 15001784.
- 1 2 Schwöbbermeyer, H (2008). "Network Motifs". In Junker BH, Schreiber F. Analysis of Biological Networks. Hoboken, New Jersey: John Wiley & Sons. pp. 85–108.
- ↑ Bornholdt, S; Schuster, HG (2003). "Handbook of graphs and networks : from the genome to the Internet". Handbook of Graphs and Networks: from the Genome to the Internet. Weinheim: Wiley-VCH. p. 417. Bibcode:2003hgnf.book.....B.
- 1 2 3 Ciriello G, Guerra C (2008). "A review on models and algorithms for motif discovery in protein-protein interaction networks". Briefings in Functional Genomics and Proteomics. 7 (2): 147–156. doi:10.1093/bfgp/eln015. PMID 18443014.
- 1 2 3 4 5 6 Kashtan N, Itzkovitz S, Milo R, Alon U (2004). "Efficient sampling algorithm for estimating sub-graph concentrations and detecting network motifs". Bioinformatics. 20 (11): 1746–1758. doi:10.1093/bioinformatics/bth163. PMID 15001476.
- 1 2 3 4 5 6 Wernicke S (2006). "Efficient detection of network motifs". IEEE/ACM Transactions on Computational Biology and Bioinformatics. 3 (4): 347–359. doi:10.1109/tcbb.2006.51.
- ↑ Picard F, Daudin JJ, Schbath S, Robin S (2005). "Assessing the Exceptionality of Network Motifs". J. Comp. Bio. 15 (1): 1–20.
- 1 2 3 Schreiber F, Schwöbbermeyer H (2005). "Frequency concepts and pattern detection for the analysis of motifs in networks". Transactions on Computational Systems Biology III: 89–104.
- 1 2 3 Shen-Orr SS, Milo R, Mangan S, Alon U (May 2002). "Network motifs in the transcriptional regulation network of Escherichia coli". Nat. Genet. 31 (1): 64–8. doi:10.1038/ng881. PMID 11967538.
- ↑ Eichenberger P, Fujita M, Jensen ST, et al. (October 2004). "The program of gene transcription for a single differentiating cell type during sporulation in Bacillus subtilis". PLOS Biology. 2 (10): e328. doi:10.1371/journal.pbio.0020328. PMC 517825
 . PMID 15383836.
. PMID 15383836. 
- ↑ Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U (October 2002). "Network motifs: simple building blocks of complex networks". Science. 298 (5594): 824–7. Bibcode:2002Sci...298..824M. doi:10.1126/science.298.5594.824. PMID 12399590.
- ↑ Lee TI, Rinaldi NJ, Robert F, et al. (October 2002). "Transcriptional regulatory networks in Saccharomyces cerevisiae". Science. 298 (5594): 799–804. Bibcode:2002Sci...298..799L. doi:10.1126/science.1075090. PMID 12399584.
- ↑ Odom DT, Zizlsperger N, Gordon DB, et al. (February 2004). "Control of pancreas and liver gene expression by HNF transcription factors". Science. 303 (5662): 1378–81. Bibcode:2004Sci...303.1378O. doi:10.1126/science.1089769. PMC 3012624. PMID 14988562.
- 1 2 Boyer LA, Lee TI, Cole MF, et al. (September 2005). "Core transcriptional regulatory circuitry in human embryonic stem cells". Cell. 122 (6): 947–56. doi:10.1016/j.cell.2005.08.020. PMC 3006442. PMID 16153702.
- ↑ Iranfar N, Fuller D, Loomis WF (February 2006). "Transcriptional regulation of post-aggregation genes in Dictyostelium by a feed-forward loop involving GBF and LagC". Dev. Biol. 290 (2): 460–9. doi:10.1016/j.ydbio.2005.11.035. PMID 16386729.
- ↑ Ma'ayan A, Jenkins SL, Neves S, et al. (August 2005). "Formation of regulatory patterns during signal propagation in a Mammalian cellular network". Science. 309 (5737): 1078–83. Bibcode:2005Sci...309.1078M. doi:10.1126/science.1108876. PMC 3032439. PMID 16099987.
- ↑ Ptacek J, Devgan G, Michaud G, et al. (December 2005). "Global analysis of protein phosphorylation in yeast". Nature. 438 (7068): 679–84. Bibcode:2005Natur.438..679P. doi:10.1038/nature04187. PMID 16319894.
- ↑ http://www.ft.unicamp.br/docentes/meira/accmotifs/
- ↑ Schreiber F, Schwobbermeyer H (2005). "MAVisto: a tool for the exploration of network motifs". Bioinformatics. 21 (17): 3572–3574. doi:10.1093/bioinformatics/bti556. PMID 16020473.
- 1 2 3 McKay BD (1981). "Practical graph isomorphism". Congressus Numerantium. 30: 45–87. arXiv:1301.1493 [cs.DM]. Bibcode:2013arXiv1301.1493M.
- 1 2 3 McKay BD (1998). "Isomorph-free exhaustive generation". Journal of Algorithms. 26 (2): 306–324. doi:10.1006/jagm.1997.0898.
- 1 2 Chen J, Hsu W, Li Lee M, et al. (2006). NeMoFinder: dissecting genome-wide protein-protein interactions with meso-scale network motifs. the 12th ACM SIGKDD international conference on Knowledge discovery and data mining. Philadelphia, Pennsylvania, USA. pp. 106–115.
- ↑ Huan J, Wang W, Prins J, et al. (2004). SPIN: mining maximal frequent sub-graphs from graph databases. the 10th ACM SIGKDD international conference on Knowledge discovery and data mining. pp. 581–586.
- ↑ Uetz P, Giot L, Cagney G, et al. (2000). "A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae". Nature. 403 (6770): 623–627. Bibcode:2000Natur.403..623U. doi:10.1038/35001009. PMID 10688190.
- 1 2 3 4 Grochow JA, Kellis M (2007). Network Motif Discovery Using Sub-graph Enumeration and Symmetry-Breaking (PDF). RECOMB. pp. 92–106. doi:10.1007/978-3-540-71681-5_7.
- 1 2 Grochow JA (2006). On the structure and evolution of protein interaction networks (PDF). Thesis M. Eng., Massachusetts Institute of Technology, Dept. of Electrical Engineering and Computer Science.
- 1 2 3 Alon N; Dao P; Hajirasouliha I; Hormozdiari F; Sahinalp S.C (2008). "Biomolecular network motif counting and discovery by color coding". Bioinformatics. 24 (13): i241–i249. doi:10.1093/bioinformatics/btn163. PMC 2718641. PMID 18586721.
- 1 2 3 4 5 Omidi S, Schreiber F, Masoudi-Nejad A (2009). "MODA: an efficient algorithm for network motif discovery in biological networks". Genes Genet Syst. 84 (5): 385–395. doi:10.1266/ggs.84.385. PMID 20154426.
- ↑ Barabasi AL, Albert R (1999). "Emergence of scaling in random networks". Science. 286 (5439): 509–512. Bibcode:1999Sci...286..509B. doi:10.1126/science.286.5439.509. PMID 10521342.
- ↑ Vázquez A, Dobrin R, Sergi D, et al. (2004). "The topological relationship between the large-scale attributes and local interaction patterns of complex networks". PNAS. 101 (52): 17940–17945. Bibcode:2004PNAS..10117940V. doi:10.1073/pnas.0406024101. PMC 539752. PMID 15598746.
- 1 2 3 4 Kashani ZR, Ahrabian H, Elahi E, Nowzari-Dalini A, Ansari ES, Asadi S, Mohammadi S, Schreiber F, Masoudi-Nejad A (2009). "Kavosh: a new algorithm for finding network motifs". BMC Bioinformatics. 10 (318): 318. doi:10.1186/1471-2105-10-318.
- ↑ Ali Masoudi-Nejad; Mitra Anasariola; Ali Salehzadeh-Yazdi; Sahand Khakabimamaghani (2012). "CytoKavosh: a Cytoscape Plug-in for Finding Network Motifs in Large Biological Networks". PLoS ONE. 7 (8): e43287. Bibcode:2012PLoSO...743287M. doi:10.1371/journal.pone.0043287. PMC 3430699. PMID 22952659.
- 1 2 3 4 Ribeiro P, Silva F (2010). G-Tries: an efficient data structure for discovering network motifs. ACM 25th Symposium On Applied Computing - Bioinformatics Track. Sierre, Switzerland. pp. 1559–1566.
- 1 2 Babu MM, Luscombe NM, Aravind L, Gerstein M, Teichmann SA (June 2004). "Structure and evolution of transcriptional regulatory networks". Current Opinion in Structural Biology. 14 (3): 283–91. doi:10.1016/j.sbi.2004.05.004. PMID 15193307.
- 1 2 Conant GC, Wagner A (July 2003). "Convergent evolution of gene circuits". Nat. Genet. 34 (3): 264–6. doi:10.1038/ng1181. PMID 12819781.
- ↑ Dekel E, Alon U (July 2005). "Optimality and evolutionary tuning of the expression level of a protein". Nature. 436 (7050): 588–92. Bibcode:2005Natur.436..588D. doi:10.1038/nature03842. PMID 16049495.
- ↑ Zabet NR (September 2011). "Negative feedback and physical limits of genes". Journal of Theoretical Biology. 248 (1): 82–91. doi:10.1016/j.jtbi.2011.06.021. PMID 21723295.
- ↑ Rosenfeld N, Elowitz MB, Alon U (November 2002). "Negative autoregulation speeds the response times of transcription networks". J. Mol. Biol. 323 (5): 785–93. doi:10.1016/S0022-2836(02)00994-4. PMID 12417193.
- ↑ Camas FM, Blázquez J, Poyatos JF (August 2006). "Autogenous and nonautogenous control of response in a genetic network". Proc. Natl. Acad. Sci. U.S.A. 103 (34): 12718–23. Bibcode:2006PNAS..10312718C. doi:10.1073/pnas.0602119103. PMC 1568915. PMID 16908855.
- ↑ Becskei A, Serrano L (June 2000). "Engineering stability in gene networks by autoregulation". Nature. 405 (6786): 590–3. doi:10.1038/35014651. PMID 10850721.
- ↑ Dublanche Y, Michalodimitrakis K, Kümmerer N, Foglierini M, Serrano L (2006). "Noise in transcription negative feedback loops: simulation and experimental analysis". Mol. Syst. Biol. 2 (1): 41. doi:10.1038/msb4100081. PMC 1681513. PMID 16883354.
- ↑ Shimoga V, White J, Li Y, Sontag E, Bleris L (2013). "Synthetic mammalian transgene negative autoregulation". Mol. Syst. Biol. 9: 670. doi:10.1038/msb.2013.27. PMC 3964311. PMID 23736683.
- ↑ Maeda YT, Sano M (June 2006). "Regulatory dynamics of synthetic gene networks with positive feedback". J. Mol. Biol. 359 (4): 1107–24. doi:10.1016/j.jmb.2006.03.064. PMID 16701695.
- ↑ Becskei A, Séraphin B, Serrano L (May 2001). "Positive feedback in eukaryotic gene networks: cell differentiation by graded to binary response conversion". EMBO J. 20 (10): 2528–35. doi:10.1093/emboj/20.10.2528. PMC 125456. PMID 11350942.
- 1 2 3 Mangan S, Alon U (October 2003). "Structure and function of the feed-forward loop network motif". Proc. Natl. Acad. Sci. U.S.A. 100 (21): 11980–5. Bibcode:2003PNAS..10011980M. doi:10.1073/pnas.2133841100. PMC 218699. PMID 14530388.
- ↑ Ma HW, Kumar B, Ditges U, Gunzer F, Buer J, Zeng AP (2004). "An extended transcriptional regulatory network of Escherichia coli and analysis of its hierarchical structure and network motifs". Nucleic Acids Res. 32 (22): 6643–9. doi:10.1093/nar/gkh1009. PMC 545451. PMID 15604458.
- ↑ Mangan S, Zaslaver A, Alon U (November 2003). "The coherent feedforward loop serves as a sign-sensitive delay element in transcription networks". J. Mol. Biol. 334 (2): 197–204. doi:10.1016/j.jmb.2003.09.049. PMID 14607112.
- ↑ Kalir S, Mangan S, Alon U (2005). "A coherent feed-forward loop with a SUM input function prolongs flagella expression in Escherichia coli". Mol. Syst. Biol. 1 (1): 2005.0006. doi:10.1038/msb4100010. PMC 1681456. PMID 16729041.
- ↑ Mangan S, Itzkovitz S, Zaslaver A, Alon U (March 2006). "The incoherent feed-forward loop accelerates the response-time of the gal system of Escherichia coli". J. Mol. Biol. 356 (5): 1073–81. doi:10.1016/j.jmb.2005.12.003. PMID 16406067.
- ↑ Entus R, Aufderheide B, Sauro HM (August 2007). "Design and implementation of three incoherent feed-forward motif based biological concentration sensors". Syst Synth Biol. 1 (3): 119–28. doi:10.1007/s11693-007-9008-6. PMC 2398716. PMID 19003446.
- ↑ Kaplan S, Bren A, Dekel E, Alon U (2008). "The incoherent feed-forward loop can generate non-monotonic input functions for genes". Mol. Syst. Biol. 4 (1): 203. doi:10.1038/msb.2008.43. PMC 2516365. PMID 18628744.
- 1 2 Bleris L, Xie Z, Glass D, Adadey A, Sontag E, Benenson Y (2011). "Synthetic incoherent feedforward circuits show adaptation to the amount of their genetic template". Mol. Syst. Biol. 7 (1): 519. doi:10.1038/msb.2011.49.
- ↑ Kalir S, McClure J, Pabbaraju K, et al. (June 2001). "Ordering genes in a flagella pathway by analysis of expression kinetics from living bacteria". Science. 292 (5524): 2080–3. doi:10.1126/science.1058758. PMID 11408658.
- ↑ Zaslaver A, Mayo AE, Rosenberg R, et al. (May 2004). "Just-in-time transcription program in metabolic pathways". Nat. Genet. 36 (5): 486–91. doi:10.1038/ng1348. PMID 15107854.
- ↑ Konagurthu AS, Lesk AM (2008). "Single and Multiple Input Modules in regulatory networks". Proteins. 73 (2): 320–324. doi:10.1002/prot.22053. PMID 18433061.
- ↑ Kaplan S, Bren A, Zaslaver A, Dekel E, Alon U (March 2008). "Diverse two-dimensional input functions control bacterial sugar genes". Mol. Cell. 29 (6): 786–92. doi:10.1016/j.molcel.2008.01.021. PMC 2366073. PMID 18374652.
- ↑ Chechik G, Oh E, Rando O, Weissman J, Regev A, Koller D (November 2008). "Activity motifs reveal principles of timing in transcriptional control of the yeast metabolic network". Nat. Biotechnol. 26 (11): 1251–9. doi:10.1038/nbt.1499. PMC 2651818. PMID 18953355.
- ↑ Ingram PJ, Stumpf MP, Stark J (2006). "Network motifs: structure does not determine function". BMC Genomics. 7: 108. doi:10.1186/1471-2164-7-108. PMC 1488845. PMID 16677373.
- ↑ Voigt CA, Wolf DM, Arkin AP (March 2005). "The Bacillus subtilis sin operon: an evolvable network motif". Genetics. 169 (3): 1187–202. doi:10.1534/genetics.104.031955. PMC 1449569. PMID 15466432.
- ↑ Knabe JF, Nehaniv CL, Schilstra MJ (2008). "Do motifs reflect evolved function?—No convergent evolution of genetic regulatory network subgraph topologies". BioSystems. 94 (1–2): 68–74. doi:10.1016/j.biosystems.2008.05.012. PMID 18611431.
- ↑ Taylor D, Restrepo JG (2011). "Network connectivity during mergers and growth: Optimizing the addition of a module". Physical Review E. 83 (6): 645–51. Bibcode:2011PhRvE..83f6112T. doi:10.1103/PhysRevE.83.066112. PMID 066112.
- ↑ Konagurthu AS, Lesk AM (2008). "On the origin of distribution patterns of motifs in biological networks". BMC Syst Biol. 2: 73. doi:10.1186/1752-0509-2-73. PMC 2538512. PMID 18700017.
External links
- Uri Alon's web page
- A software tool that can detect network motifs
- bio-physics-wiki NETWORK MOTIFS
- FANMOD: a tool for fast network motif detection
- MAVisto: network motif analysis and visualisation tool
- NeMoFinder
- Grochow-Kellis
- Noga Alon's web page
- MODA
- Kavosh
- CytoKavosh
- G-Tries
- acc-MOTIF detection tool