Duocarmycin
The duocarmycins are members of a series of related natural products first isolated from Streptomyces bacteria in 1988.[1][2][3] They are notable for their extreme cytotoxicity and thus represent a class of exceptionally potent antitumour antibiotics.[4][5]
Biological activity
As small-molecule, synthetic, DNA minor groove binding alkylating agents, duocarmycins are suitable to target solid tumors. They bind to the minor groove of DNA and alkylate the nucleobase adenine at the N3 position.[6][7] The irreversible alkylation of DNA disrupts the nucleic acid architecture, which eventually leads to tumor cell death. Analogues of naturally occurring antitumour agents, such as duocarmycins, represent a new class of highly potent antineoplastic compounds.[8][9]
The work of Dale L. Boger and others created a better understanding of the pharmacophore and mechanism of action of the duocarmycins. This research has led to synthetic analogs including adozelesin, bizelesin, and carzelesin which progressed into clinical trials for the treatment of cancer. Similar research that Boger utilized for comparison to his results involving elimination of cancerous tumors and antigens was centered around the use of similar immunoconjugates that were introduced to cancerous colon cells. These studies related to Boger's research involving antigen-specificity that is necessary to the success of the duocarmycins as antitumor treatments.[10]
Duocarmycin analogues vs tubulin binders
The duocarmycin analogues are able to exert their mode of action at any phase in the cellular cycle, whereas tubulin binders will only attack tumor cells when they are in a mitotic state. Growing evidence suggests that DNA damaging agents, such as duocarmycins, are more efficacious in tumor cell killing than tubulin binders, particularly in case of solid tumors. They have shown activity in a variety of multi-drug resistant (MDR) models. Agents that are part of this class of duocarmycins have the potency in the low picomolar range. This makes them suitable for maximizing the cell-killing potency of antibody-drug conjugates to which they are attached. Another important benefit is that, unlike other drug classes, duocarmycins can be effective against tumor cells that are multi-drug resistant. For example, potent cytotoxicity has been demonstrated in cells that express the P-glycoprotein (P-gp) efflux pump. Multi-drug resistance presents a significant problem in the clinical setting and agents that are less susceptible to these mechanisms can successfully be used in prolonged treatment protocols.[11]
Duocarmycins
-
-Duocarmycin_A.svg.png)
Duocarmycin A
-
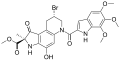
Duocarmycin B1
-
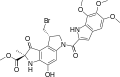
Duocarmycin B2
-
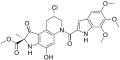
Duocarmycin C1
-
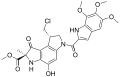
Duocarmycin C2
-
Duocarmycin D
-
Duocarmycin SA
-
CC-1065
Antibody-drug conjugates
The DNA modifying agents such as duocarmycin are being used in the development of antibody-drug conjugate or ADCs. Scientists at The Netherlands-based Synthon (formerly Syntarga) have combined a unique linkers with duocarmycin derivatives that have a hydroxyl group which is crucial for biological activity. Using this technology scientists aims to create ADCs having an optimal therapeutic window, balancing the effect of potent cell-killing agents on tumor cells versus healthy cells.[12]
Synthetic analogs
The synthetic analogs of duocarmycins include adozelesin, bizelesin, and carzelesin. As members of the cyclopropylpyrroloindole family, these investigational drugs have progressed into clinical trials for the treatment of cancer.
Adozelesin
Adozelesin is an alkylating minor groove DNA binder that is capable of rapidly inhibiting DNA replication in treated cells through a trans-acting mechanism and preferentially arrests cells in S phase. It has been shown previously that in cells treated with adozelesin, replication protein A (RPA) activity is deficient, and the middle subunit of RPA is hyperphosphorylated. The adozelesin-induced RPA hyperphosphorylation can be blocked by the replicative DNA polymerase inhibitor, aphidicolin, suggesting that adozelesin-triggered cellular DNA damage responses require active DNA replication forks. These data imply that cellular DNA damage responses to adozelesin treatment are preferentially induced in S phase.[13]
Bizelesin
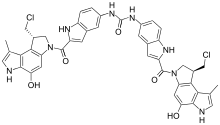
Bizelesin is antineoplastic antibiotic which binds to the minor groove of DNA and induces interstrand cross-linking of DNA, thereby inhibiting DNA replication and RNA synthesis. Bizelesin also enhances p53 and p21 induction and triggers G2/M cell-cycle arrest, resulting in cell senescence without apoptosis.[14]
Adozelesin and bizelesin cause genomic DNA lesions by alkylating DNA. Adozelesin induces single-strand DNA lesions, whereas bizelesin induces both single-strand lesions and double-strand DNA cross-links. At equivalent cytotoxic concentrations, these agents caused different biological responses. Low adozelesin concentrations (e.g., 0.5 nM) induced a transient S-phase block and cell cycle arrest in G(2)-M, as well as increased induction of p53 and p21, whereas a high drug concentration (e.g., 2.5 nM) caused apoptosis but no p21 induction. In contrast, both low and high bizelesin concentrations enhanced p53 and p21 induction and triggered G(2)-M cell cycle arrest and eventual senescence without significant apoptotic cell death. However, in cells lacking p21, bizelesin, as well as adozelesin, triggered apoptosis, indicating that p21 was crucial to sustained bizelesin-induced G(2)-M arrest. Despite similar abilities to alkylate DNA, adozelesin and bizelesin caused a decrease in HCT116 tumor cell proliferation by different pathways.[15]
Carzelesin
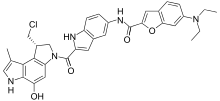
Carzelesin (U-80244) is a cyclopropylpyrroloindole prodrug containing a relatively nonreactive chloromethyl precursor to the cyclopropyl function. Activation of carzelesin requires two steps, (a) hydrolysis of a phenylurethane substituent to form U-76073, followed by (b) ring closure to form the cyclopropyl-containing DNA-reactive U-76074. The formation of the DNA-reactive U-76074, via U-76073, from carzelesin was shown to proceed very slowly in phosphate-buffered saline (t1/2 greater than 24 h) but to occur rapidly in plasma from mouse, rat, dog, and human (initial t1/2 values ranging from 18 min for mouse to 52 min for rat) and in cell culture medium (t1/2 approximately 40 min). Although carzelesin was less potent in terms of in vitro cytotoxicity and in vivo optimal dosage and showed low affinity for binding to DNA, it was therapeutically more efficacious against mouse L1210 leukemia than was U-76074 or adozelesin (U-73975), another cyclopropylpyrroloindole analogue which is currently in phase I clinical trials. Carzelesin also proved to be more efficacious than U-76074 or adozelesin against mouse pancreatic ductal 02 adenocarcinoma, a system reported to be resistant to every agent tested. Carzelesin was highly effective against this tumor and produced 97% tumor growth inhibition. In addition, i.v. administered carzelesin showed significant activity (National Cancer Institute criteria) against i.v. or s.c. implanted Lewis lung carcinoma, i.p. or s.c. implanted B16 melanoma, s.c. implanted colon 38 carcinoma, and five s.c. implanted human tumor xenografts, including clear cell Caki-1 carcinoma, colon CX-1 adenocarcinoma, lung LX-1 tumor, ovarian 2780 carcinoma, and prostatic DU-145 carcinoma. Carzelesin treatment produced 100% complete remissions (no palpable tumor mass at the termination of the experiment) in mice bearing early-stage human ovarian 2780. Pharmacologically, carzelesin proved to be relatively schedule and route independent and was highly active against i.p. implanted L1210 leukemia, regardless of whether the analogue was given i.v., i.p., s.c., or p.o. These results, collectively, suggest that carzelesin is absorbed and distributed well. Both carzelesin and adozelesin caused marked tumor shrinkage in mice bearing human lung LX-1 or advanced-stage human ovarian 2780 carcinoma; however, tumor regrowth occurred shortly after the treatment with adozelesin was stopped. Little or no apparent tumor regrowth occurred after treatment with carzelesin.[16]
References
- ↑ Yasuzawa, Tohru; Iida, Takao; Muroi, Ken'Ichi; Ichimura, Michio; Takahashi, Keiichi; Sano, Hiroshi (1988). "Structures of Duocarmycins, novel antitumor antibiotics produced by Streptomyces sp". Chemical & Pharmaceutical Bulletin. 36 (9): 3728–31. doi:10.1248/cpb.36.3728.
- ↑ Takahashi, Isami; Takahashi, KEI-Ichi; Ichimura, Michio; Morimoto, Makoto; Asano, Kozo; Kawamoto, Isao; Tomita, Fusao; Nakano, Hirofumi (1988). "Duocarmycin A, a new antitumor antibiotic from Streptomyces". The Journal of Antibiotics. 41 (12): 1915–7. doi:10.7164/antibiotics.41.1915. PMID 3209484.
- ↑ "Cytotoxic Agents". ADC Review/Journal of Antibody-drug Conjugates. October 29, 2013.
- ↑ Boger, Dale L. (1991). "Duocarmycins: a new class of sequence selective DNA minor groove alkylating agents". Chemtracts: Organic Chemistry. 4 (5): 329–49.
- ↑ Tercel, Moana; McManaway, Sarah P.; Leung, Euphemia; Liyanage, H. D. Sarath; Lu, Guo-Liang; Pruijn, Frederik B. (2013). "The Cytotoxicity of Duocarmycin Analogues is Mediated through Alkylation of DNA, not Aldehyde Dehydrogenase 1: A Comment". Angewandte Chemie International Edition. 52 (21): 5442–6. doi:10.1002/anie.201208373.
- ↑ Boger, D. L. (1993). "Design, synthesis, and evaluation of DNA minor groove binding agents". Pure and Applied Chemistry. 65 (6): 1123–32. doi:10.1351/pac199365061123.
- ↑ Boger, Dale L.; Johnson, Douglas S. (1995). "CC-1065 and the Duocarmycins: Unraveling the Keys to a New Class of Naturally Derived DNA Alkylating Agents". Proceedings of the National Academy of Sciences of the United States of America. 92 (9): 3642–9. Bibcode:1995PNAS...92.3642B. doi:10.1073/pnas.92.9.3642. PMC 42018
 . PMID 7731958.
. PMID 7731958. - ↑ Tietze, Lutz F.; Krewer, Birgit (2009). "Antibody-Directed Enzyme Prodrug Therapy: A Promising Approach for a Selective Treatment of Cancer Based on Prodrugs and Monoclonal Antibodies". Chemical Biology & Drug Design. 74 (3): 205–11. doi:10.1111/j.1747-0285.2009.00856.x.
- ↑ Cacciari, Barbara; Romagnoli, Romeo; Baraldi, Pier Giovanni; Ros, Tatiana Da; Spalluto, Giampiero (2000). "CC-1065 and the duocarmycins: Recent developments". Expert Opinion on Therapeutic Patents. 10 (12): 1853–71. doi:10.1517/13543776.10.12.1853.
- ↑ Liu, C; Tadayoni, B M; Bourret, L A; Mattocks, K M; Derr, S M; Widdison, W C; Kedersha, N L; Ariniello, P D; Goldmacher, V S (1996-08-06). "Eradication of large colon tumor xenografts by targeted delivery of maytansinoids.". Proceedings of the National Academy of Sciences of the United States of America. 93 (16): 8618–8623. ISSN 0027-8424. PMC 38722. PMID 8710920.
- ↑ "Duocarmycin analogues". ADC Review/Journal of Antibody-drug Conjugates. November 26, 2014.
- ↑ Hofland, Peter. "First-in-Human Trial With SYD985 Evaluates Safety and Efficacy in Cancer Patients". ADCReview /Journal of Antibody-drug Conjugates (November 2014).
- ↑ Liu, Jen-Sing; Kuo, Shu-Ru; Beerman, Terry A.; Melendy, Thomas (2003). "Induction of DNA Damage Responses by Adozelesin Is S Phase-specific and Dependent on Active Replication Forks". Molecular Cancer Therapeutics. 2 (1): 41–7. PMID 12533671.
- ↑ "Bizelesin". MedKoo Biosciences.
- ↑ Cao, Pei-rang; McHugh, Mary M.; Melendy, Thomas; Beerman, Terry (2003). "The DNA Minor Groove-alkylating Cyclopropylpyrroloindole Drugs Adozelesin and Bizelesin Induce Different DNA Damage Response Pathways in Human Colon Carcinoma HCT116 Cells". Molecular Cancer Therapeutics. 2 (7): 651–9. PMID 12883038.
- ↑ Li, L. H.; Dekoning, T. F.; Kelly, R. C.; Krueger, W. C.; McGovren, J. P.; Padbury, G. E.; Petzold, G. L.; Wallace, T. L.; Ouding, R. J.; Prairie, M. D. (1992). "Cytotoxicity and Antitumor Activity of Carzelesin, a Prodrug Cyclopropylpyrroloindole Analogue". Cancer Research. 52 (18): 4904–13. PMID 1516047.