Cieplak Effect
In organic chemistry, the Cieplak effect is a predictive model that explains why nucleophiles preferentially add to one face of a carbonyl over another. Proposed by Andrzej Stanislaw Cieplak in 1980, it explains anomalous results that other models of the time, such as the Cram and Felkin-Anh models, can't justify. In the Cieplak model, electrons from a neighboring bond delocalize into the forming carbon-nucleophile (C-Nuc) bond, lowering the energy of the transition state and accelerating the rate of reaction.[1] Whichever bond can best donate its electrons into the C-Nuc bond determines which face of the carbonyl the nucleophile will add to. The nucleophile may be a number of reagents, most commonly organometallic or reducing agents. The Cieplak effect is subtle, and often competes with sterics, solvent effects, counterion complexation of the carbonyl oxygen, and other effects to determine product distribution.
Background
The Cieplak effect relies on the stabilizing interaction of mixing full and empty orbitals to delocalize electrons, known as hyperconjugation.[2] When the highest occupied molecular orbital (HOMO) of one system and the lowest unoccupied molecular orbital (LUMO) of another system have comparable energies and spatial overlap, the electrons can delocalize and sink into a lower energy level. Often, the HOMO of a system is a full σ (bonding) orbital and the LUMO is an empty σ* (antibonding) orbital. This mixing is a stabilizing interaction and has been widely used to explain such phenomena as the anomeric effect. A common requirement of hyperconjugation is that the bonds donating and accepting electron density are antiperiplanar to each other, to allow for maximum orbital overlap.
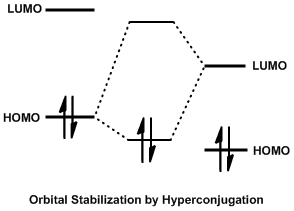
The Cieplak effect uses hyperconjugation to explain the face-selective addition of nucleophiles to carbonyl carbons. Specifically, donation into the low-lying σ*C-Nuc bond by antiperiplanar electron-donating substituents is the stabilizing interaction which lowers the transition state energy of one stereospecific reaction pathway and thus increases the rate of attack from one side. In the simplest model, a conformationally constrained cyclohexanone is reduced to the corresponding alcohol. Reducing agents add a hydride to the carbonyl carbon via attack along the Burgi-Dunitz angle, which can come from the top along a pseudo-axial trajectory or from below, along a pseudo-equatorial trajectory. It has long been known that large reducing agents add hydride to the equatorial position to avoid steric interactions with axial hydrogens on the ring. Small hydride sources, however, add hydride to an axial position for reasons which are still disputed.[3]
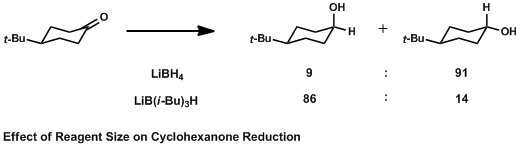
The Cieplak effect explains this phenomenon by postulating that hyperconjugation of the forming σ*C-H orbital with geometrically aligned σ orbitals is the stabilizing interaction that controls stereoselectivity. In an equatorial approach, the bonds that are geometrically aligned antiperiplanar to the forming C-H bond are the C-C bonds of the ring, so they donate electron density to σ*C-H. In an axial approach, the neighboring axial C-H bonds are aligned antiperiplanar to the forming C-H bond, so they donate electron density to σ*C-H. Because C-H bonds are better electron donors than C-C bonds, they are better able to participate in this stabilizing interaction and so this pathway is favored.[1]

Evidence
Cieplak’s proposal is supported by investigating the effects of various electronic substituents on product distribution. By installing an electron-withdrawing substituent such as a methoxy group at the C2 position, the reduction of substituted cyclohexanones begins to favor equatorial attack.[4] This is because the axial C-O bond is a worse electron donor than a C-C bond, so axial attack is less favored.

Cieplak also demonstrated this effect by introducing electron withdrawing substituents on C3, which decrease the electron-donating capability of the ring C-C bond and therefore disfavor equatorial attack, which is antiperiplanar to this bond.[5] Electron-donating substituents at C3 subsequently favor equatorial approach, since increasing C-C electron density favors σC-C donation into σ*C-Nuc and thereby encourages equatorial approach.

This effect can also be investigated by changing the electronic nature of the nucleophile. In the case of an electron-deficient nucleophile, the σ* of the forming C-Nuc bond is lower in energy and better stabilized by attack antiperiplanar to electron-rich axial C-H bonds. Attack therefore occurs axially.[6] If the nucleophile is electron-rich, however, the donation of more electron density is less favored and equatorial attack may prevail.[7] These trends have been observed even when normalizing for steric bulk of the nucleophile.[1]
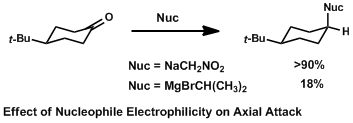
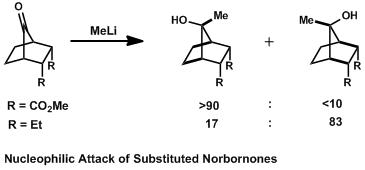
In substituted norbornones, nucleophilic attack will come antiperiplanar to the bonds which can best donate into σ*C-Nuc.[8] The bonds positioned for this interaction are the bridgehead C-C bonds on the six-membered ring. Substituents which donate electron density to these bonds, such as ethyl groups, increase the rate of addition anti to the alkyl groups, which is the antiperiplanar trajectory. If electron-withdrawing substituents such as esters are appended to the C-C bonds, however, the selectivity favors syn addition, so that the bonds donating into σ*C-Nuc are the more electron-rich C-C bonds which are hydrogen-substituted.[9]
A similar example is seen in substituted 2-adamantones, where varying the electronic properties at the remote 5 position has profound effects on product distribution.[10] A hydroxyl group is able to donate electron density inductively to the forming σ*C-H bond antiperiplanar, so attack from that side is favored. The electron-withdrawing ester substituent, however, lacks this stabilization ability. Instead, the C-H bonds are better electron donors than the C-CO2Me bonds, so attack comes anti to the hydrogen substituents and subsequently syn to the ester group. This explains the effect of remote electron-donor groups on stereochemical outcomes, which has been difficult to explain with other stereochemical models. The rigidity of the adamantone skeleton allows for tight control of conformation and minimization of competing effects.[11]
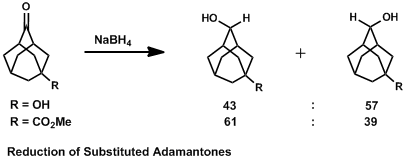
Criticisms
The Cieplak model has been met with mixed reviews, and criticisms of both its basic logic and predictive ability have emerged. The stabilizing interaction of donating electron density into the transition state σ* orbital of a forming bond was immediately questioned, as this interaction has been widely invoked to explain exactly the opposite—the destabilization of bonds.[12] Traditionally, forming bonds are stabilized when they donate electron density from their bonding HOMO into a neighboring antibonding LUMO, not by accepting electron density into their LUMO. To this end, David A. Evans said of Cieplak’s proposal: "Structures are stabilized by stabilizing their highest energy filled states. This is one of the fundamental assumptions in frontier molecular orbital theory. The Cieplak hypothesis is nonsense."[13] However, Hahn and le Noble refute this point by invoking the principle of microscopic reversibility, where the process of bond formation and cleavage are fundamentally equivalent in an equilibrium, and little value should be placed on the terms ‘bonding’ and ‘antibonding’, σ or σ*.[14] In another criticism of the model, Houk questions Cieplak’s fundamental assumption that C-H bonds are better electron-donors than C-C bonds.[15] This point is still contested and represents another major dispute in the field.
To further refute the Cieplak effect, Houk puts forth a system in which Cieplak’s model predicts the wrong product distribution trend. In the case of substituted trans-decalones, electron-withdrawing substituents equatorial at C4 should discourage equatorial attack and yield more axial product, since the ring C-C bonds are deactivated for donation into the forming C-Nuc bond. Experimental evidence, however, shows that axial electron-withdrawing C4 substituents are more directing towards axial attack than equatorial substituents.[16] Since axial orbitals are not aligned for hyperconjugation in this system, Houk rationalized this trend by invoking electrostatic arguments, described below.
Alternative explanations
In an effort to explain the surprising stereoselectivies in the systems above, alternative explanations to the Cieplak effect have been proposed. In substituted cyclohexanones, the tendency of small reducing agents to add hydride axially is proposed to be caused by torsional strain instead of hyperconjugation.[17] In an equatorial attack, the nucleophile approaches by eclipsing a neighboring hydrogen atom and subsequently pushes the carbonyl subsituents into ecplising positions as it pyramidalizes the carbonyl carbon.[18] In an axial approach, the nucleophile approaches gauche to neighboring hydrogen atoms and so does not cause eclipsing interactions while pyramidalizing the carbonyl carbon. It is this torsional strain—the energy cost of rotating the bonds out of an eclipsed position—that favors axial over equatorial approach.
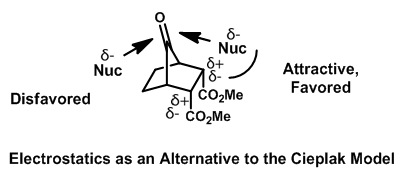
In the case of substituted norbornones, stereoselectivity may be explained by electrostatic interactions between substituents and nucleophiles. Electron-withdrawing groups create a partial positive charge on the alpha carbon, which interacts favorably with the partial negative charge on the incoming nucleophile.[19] This interaction may guide attack syn to the electron-withdrawing substituent, and anti to electron-donating substituents. This conclusion is supported by computations, where modeling the partial charges predicts product distribution without including orbital interactions.[20] The same explanation has been made to justify similar results in the case of substituted adamantones.[21]
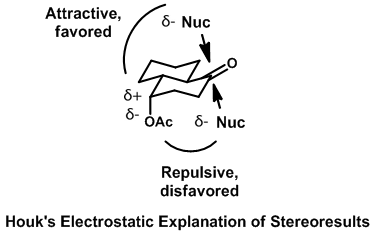
Similarly, in Houk’s trans-decalone system, the nucleophile with its partial negative charge prefers to attack away from the partial negative charge of the acyl ester. When this substituent is axial, the equatorial pathway brings the nucleophile into closer proximity and is therefore disfavored.[22] This is less pronounced for equatorially substituted ester because the group is now positioned further away from the carbonyl.[16]
While the validity of the Cieplak effect is still disputed, it may be the best model to explain certain anomalous stereochemical results. As chemists continue to discover new reactions and conditions, we continually test, disprove, and propose new models. As such, it is important to continue debating and discussing theoretical frameworks for the physical phenomena we encounter, in the hope of one day getting it right.
References
- 1 2 3 Cieplak, A. S. J. Am. Chem. Soc. 1981, 103, 4540
- ↑ Alabugin, I. V.; Gilmore, K. M.; Peterson, P. W. WIREs Comput. Mol. Sci. 2011, 1, 109
- ↑ Eliel, E. L.; Senda, Y. Tetrahedron 1970, 26, 2411
- ↑ Senda, Y.; Nakano, S.; Kunii, H.; Itoh, H. J. Chem. Soc. Perkin Trans. 1993, 2, 1009
- ↑ Johnson, C. R.; Tait, B. D.; Cieplak, A. S. J. Am. Chem. Soc. 1987, 109, 5875
- ↑ Favre, H.; Gravel, D. Can. J. Chem. 1961, 39, 1548
- ↑ Meakins, G. D.; Percy, R. K.; Richards, E. E.;Young, R. N. J. Chem. Soc. C, 1968, 1106
- ↑ G. Mehta, J. Am. Chem. Soc. 1990, 112, 6140
- ↑ Kaselj, M.; Chung, W-S.; le Noble, W. J. Chem. Rev. 1999, 99, 1387
- ↑ Cheung, C. K.; Tseng, L. T.; Lin, M. H.; Srivsstava, S.; le Noble, W. J. J. Am. Chem. Soc. 1986, 108, 1598
- ↑ Srivastava, S.; le Noble, W. J. J. Am. Chem. Soc. 1987, 109, 5874
- ↑ Frenking, G.; Kohler, K. F.; Reetz, M. T. Angew. Chem. Int. Ed. Engl. 1991, 30, 1146
- ↑ Siska, S. (2001, February). The Evolution of Models for Carbonyl Addition. Evans Group Afternoon Seminar. Lecture conducted from Harvard University, Cambridge, MA
- ↑ Hahn, J. M.; le Noble. W. J. J. Am. Chem. Soc. 1992, 114, 1916
- ↑ Rozeboom, M. D.; Houk, K. N. J. Am. Chem. Soc. 1982, 104, 1189
- 1 2 Wu, Y-D.; Tucker, J. A.; Houk, K. N. J. Am. Chem. Soc. 1991, 113, 5018
- ↑ Juaristi, E. Conformational Behavior of Six-Membered Rings. Wiley, 1995. Online.
- ↑ Wigfield, D. C. Tetrahedron 1979, 35, 449
- ↑ Paddon-Row, M. N.; Wu, Y-D.; Houk, K. N. J. Am. Chem. Soc. 1992, 114, 10638
- ↑ Ganguly, B.; Chandrasekhar, J.; Khan, F. A.; Mehta, G. J. Org. Chem. 1993, 58, 1734
- ↑ Adcock, W.; Cotton, J.; Trout, N. A. J. Org. Chem. 1994, 59, 1867
- ↑ Northrup, A. B. (2003, September). Hyperconjugation. MacMillan Group Meeting. Lecture conducted from University of California, Bekeley, CA.